GASTROENTEROLOGY
1. Gastroesophageal reflux
disease. Etiology and pathogenesis. Clinical forms.
is a chronic condition of mucosal damage caused bystomach acid coming up from the stomach into the esophagus
is a chronic condition of mucosal damage caused bystomach acid coming up from the stomach into the esophagus
Gastro
esophageal reflux disease
Etiology
Abnormalities of the lower oesophageal sphincter
Hiatus hernia: Herniation of the stomach through
the diaphragm into the chest
Delayed oesophageal clearance
Gastric contents
Defective gastric emptying
Increased intra-abdominal pressure
Dietary and environmental factors
Patient
factors: Visceral
sensitivity

2.
The different types of GERD include:
2. Symptoms of gastroesophageal reflux disease. Instrumental and laboratory methods of diagnostics. Principles of treatment.
The different types of GERD include:
- Gastroesophageal Reflux Disease (GERD)
Gastroesophageal reflux disease (GERD), also known as acid reflux disease, occurs when the lower esophageal sphincter (LES) does not close properly and stomach contents leak back, or reflux, into the esophagus. - Refractory Gastroesophageal Reflux Disease (Refractory GERD)
The term refractory gastroesophageal reflux disease (refractory GERD) describes those patients who continue to have symptoms of gastroesophageal reflux despite standard treatment with proton pump inhibitors (PPIs). - Nonerosive Reflux Disease (NERD)
For some patients, GERD can cause erosive esophagitis, a condition that causes inflammation, swelling, or irritation of the esophagus. In recent studies, however, it has been found that less than half of GERD patient suffer from esophagitis. They have what is called nonerosive reflux disease, or NERD. - Larygopharyngeal Reflux (LPR)
When the lower esophageal sphincter (LES) is not functioning properly, there is a back flow of stomach acid into the esophagus. If this happens two or more times a week, it can be a sign of gastroesophageal reflux disease, or GERD. As with the lower esophageal sphincter, if the upper esophageal sphincter doesn't function properly, acid that has back flowed into the esophagus is allowed into the throat and voice box. When this happens, it's called Laryngopharyngeal Reflux, or LPR.
2. Symptoms of gastroesophageal reflux disease. Instrumental and laboratory methods of diagnostics. Principles of treatment.
The major symptoms are
heartburn and egurgitation, often provoked by bending, straining or lying down.
‘Waterbrash’, which is salivation due to reflex salivary gland stimulation as
acid enters the gullet, is often present. The patient is often overweight. Some
patients are woken at night by choking as refluxed fluid irritates the larynx.
Others develop odynophagia or dysphagia. A variety of other features have been
described, such as atypical chest pain which may be severe and can mimic
angina, and may be due to reflux-induced oesophageal spasm. Others include
hoarseness (‘acid laryngitis’), recurrent chest infections, chronic cough and
asthma. The true relationship of these features to gastro-oesophageal reflux
disease remains unclear.
diagnosis
1. A presumptive
diagnosis of GERD can be established in the setting of typical symptoms of
heartburn and regurgitation. Empiric medical therapy with a proton pump
inhibitor (PPI) is recommended in this setting. (Strong recommendation,
moderate level of evidence)
2. Patients with
non-cardiac chest pain suspected due to GERD should have diagnostic evaluation
before institution of therapy. (Conditional recommendation, moderate level of
evidence). A cardiac cause should be excluded in patients with chest pain
before the commencement of a gastrointestinal evaluation (Strong
recommendation, low level of evidence)
3. Barium radiographs
should not be performed to diagnose GERD (Strong recommendation, high level of
evidence)
4. Upper endoscopy is
not required in the presence of typical GERD symptoms. Endoscopy is recommended
in the presence of alarm symptoms and for screening of patients at high risk
for complications. Repeat endoscopy is not indicated in patients without
Barrett’s esophagus in the absence of new symptoms. (Strong recommendation,
moderate level of evidence)
5. Routine biopsies
from the distal esophagus are not recommended specifically to diagnose GERD.
(Strong recommendation, moderate level of evidence)
6. Esophageal
manometry is recommended for preoperative evaluation, but has no role in the
diagnosis of GERD. (Strong recommendation, low level of evidence)
7. Ambulatory esophageal
reflux monitoring is indicated before consideration of endoscopic or surgical
therapy in patients with non-erosive disease, as part of the evaluation of
patients refractory to PPI therapy, and in situations when the diagnosis of
GERD is in question. (Strong recommendation, low level of evidence). Ambulatory
reflux monitoring is the only test that can assess reflux symptom association
(strong recommendation, low level of evidence).
8. Ambulatory reflux
monitoring is not required in the presence of short or long-segment Barrett’s
esophagus to establish a diagnosis of GERD. (Strong recommendation, moderate
level of evidence)
9. Screening for
Helicobacter pylori infection is not recommended in GERD patients. Treatment of
H. pylori infection is not routinely required as part of antireflux therapy.
(Strong recommendation, low level of evidence)
therapy
1. Weight loss is
recommended for GERD patients who are overweight or have had recent weight
gain. (Conditional recommendation, moderate level of evidence)
2. Head of bed
elevation and avoidance of meals 2–3 h before bedtime should be recommended for
patients with nocturnal GERD. (Conditional recommendation, low level of
evidence)
3. Routine global
elimination of food that can trigger reflux (including chocolate, caffeine,
alcohol, acidic and/or spicy foods) is not recommended in the treatment of
GERD. (Conditional recommendation, low level of evidence)
4. An 8-week course of
PPIs is the therapy of choice for symptom relief and healing of erosive esophagitis.
There are no major differences in efficacy between the different PPIs. (Strong
recommendation, high level of evidence)
5. Traditional delayed
release PPIs should be administered 30–60 min before meal for maximal pH
control. (Strong recommendation, moderate level of evidence). Newer PPIs may
offer dosing flexibility relative to meal timing. (Conditional recommendation,
moderate level of evidence)
6. PPI therapy should
be initiated at once a day dosing, before the first meal of the day. (Strong
recommendation, moderate level of evidence). For patients with partial response
to once daily therapy, tailored therapy with adjustment of dose timing and/or
twice daily dosing should be considered in patients with night-time symptoms,
variable schedules, and/or sleep disturbance. (Strong recommendation, low level
of evidence).
7. Non-responders to
PPI should be referred for evaluation. (Conditional recommendation, low level
of evidence, see refractory GERD section).
8. In patients with
partial response to PPI therapy, increasing the dose to twice daily therapy or
switching to a different PPI may provide additional symptom relief.
(Conditional recommendation, low level evidence).
9. Maintenance PPI
therapy should be administered for GERD patients who continue to have symptoms
after PPI is discontinued, and in patients with complications including erosive
esophagitis and Barrett’s esophagus. (Strong recommendation, moderate level of
evidence). For patients who require long-term PPI therapy, it should be
administered in the lowest effective dose, including on demand or intermittent
therapy. (Conditional recommendation, low level of evidence)
10. H2-receptor
antagonist (H2RA) therapy can be used as a maintenance option in patients
without erosive disease if patients experience heartburn relief. (Conditional
recommendation, moderate level of evidence). Bedtime H2RA therapy can be added
to daytime PPI therapy in selected patients with objective evidence of
night-time reflux if needed, but may be associated with the development of
tachyphlaxis after several weeks of use. (Conditional recommendation, low level
of evidence)
11. Therapy for GERD
other than acid suppression, including prokinetic therapy and/or baclofen,
should not be used in GERD patients without diagnostic evaluation. (Conditional
recommendation, moderate level of evidence)
12. There is no role
for sucralfate in the non-pregnant GERD patient. (Conditional recommendation,
moderate level of evidence)
13. PPIs are safe in
pregnant patients if clinically indicated. (Conditional recommendation,
moderate level of evidence)
Surgical options for
GERD
1. Surgical therapy is
a treatment option for long-term therapy in GERD patients. (Strong
recommendation, high level of evidence)
2. Surgical therapy is
generally not recommended in patients who do not respond to PPI therapy.
(Strong recommendation, high level of evidence)
3. Preoperative
ambulatory pH monitoring is mandatory in patients without evidence of erosive
esophagitis. All patients should undergo preoperative manometry to rule out
achalasia or scleroderma-like esophagus. (Strong recommendation, moderate level
of evidence)
4. Surgical therapy is
as effective as medical therapy for carefully selected patients with chronic
GERD when performed by an experienced surgeon. (Strong recommendation, high
level of evidence)
5. Obese patients
contemplating surgical therapy for GERD should be considered for bariatric
surgery. Gastric bypass would be the preferred operation in these patients.
(Conditional recommendation, moderate level of evidence)
6. The usage of
current endoscopic therapy or transoral incisionless fundoplication cannot be
recommended as an alternative to medical or traditional surgical therapy.
(Strong recommendation, moderate level of evidence)
3. Functional gastric dyspepsia.
Etiology and pathogenesis. Classification.
Etiology
Classification
Functional dyspepsia (FD) is a
chronic disorder of sensation and movement (peristalsis) in the upper digestive
tract.
Non-ulcer dyspepsia is
sometimes called functional dyspepsia. It means that no known cause can be
found for the symptoms.
The cause of functional
dyspepsia is unknown; however, several hypotheses could explain this condition
even though none can be consistently associated with FD. Excessive acid
secretion, inflammation of the stomach or duodenum, food allergies, lifestyle
and diet influences, psychological factors, medication side effects (from drugs
such as non-steroidal anti-inflammatory drugs and aspirin), and Helicobacter pylori infection have all had their proponents.
Classification
Postprandial Distress Syndrome
Epigastric Pain Syndrome
4. Functional gastric dyspepsia.
Principles of treatment.
Dietary and Lifestyle Modifications
Medications
H2 Receptor Antagonists: cimetidine (Tagamet®), ranitidine (Zantac®), famotidine (Pepcid®), and nizatidine (Axid®).
PPIs: omeprazole (Losec®), lansoprazole (Prevacid®), pantoprazole sodium (Pantoloc®), esomeprazole (Nexium®), rabeprazole (Pariet®), and pantoprazole magnesium (Tecta®).
PPIs: omeprazole (Losec®), lansoprazole (Prevacid®), pantoprazole sodium (Pantoloc®), esomeprazole (Nexium®), rabeprazole (Pariet®), and pantoprazole magnesium (Tecta®).
5. Chronic gastritis. Etiology and
pathogenesis. Classification (Sydney).
Etiology
Causes: Chronic
gastritis is polyetiologic disease. The main causes of chronic gastritis are:
1) Infection -
Helicobacter pylori.
2) Immunologic factors,
which lead to the production of the autoantibodies to the parietal cells.
3) Chemical factors,
which lead to the injury of the gastric mucosa (alcohol, aspirin and other
NSAIDs, reflux of bile and pancreatic secretions).
4) Radiation or
thermal injury.
Factors, which promote
to the development of the chronic gastritis, are exogenous and endogenous.
Exogenous factors are:
1) abnormality of the
order of nutrition, overeating, insufficient chewing of food, abuse of hot and
spicy food;
2) smoking and
alcohol;
3) professional harm
(swallowing of metallic dust or other chemical substances);
4) prolonged intake of
some medicines (aspirin and other
NSAIDs,
corticosteroids).
Endogenous factors
are:
1) chronic infections,
2) endocrine diseases,
3) metabolic
disorders,
4) autointoxication
(renal failure).
Pathogenesis
Atrophic gastritis is
associated with serum antiparietal and anti-intrinsic factor antibodies.
Cell-mediated immunity also contributes to the disease. T lymphocytes
infiltrate the gastric mucosa and contribute to epithelial cell destruction and
resulting gastric atrophy. Atrophic gastritis involves the fundus and the body
of the stomach. With progressive degeneration of the mucosa, gastric secretion
progressively fails. Hydrochloric acid secretion fails first, followed by
pepsinogen secretion and finally by the secretion of intrinsic factor. As
intrinsic factor secretion fails, the body becomes depleted of vitamin B12 and
pernicious anemia develops.
Helicobacter pylori (H. pylory)
are gram-negative rods that have
the ability to colonize and
infect the stomach. The bacteria survive
within the mucous layer that
covers the gastric surface epithelium and
the upper portions of the
gastric foveolae. The infection usually is
acquired during childhood. Once
the organism has been acquired, has
passed through the mucous layer,
and has become established at the
luminal surface of the stomach,
an intense inflammatory response of
the underlying tissue develops.
Previously this type of gastritis was
known as type B gastritis.
Chemical gastritis is caused by
injury of the gastric mucosa by
reflux of bile and pancreatic
secretions into the stomach, but it can
also be caused by exogenous
substances, including NSAIDs,
acetylsalicylic acid,
chemotherapeutic agents, and alcohol. These
chemicals cause epithelial
damage, erosions, and ulcers that are
followed by regenerative
hyperplasia, histologically detectable as
foveolar hyperplasia and damage
to capillaries, with mucosal edema,
hemorrhage, and proliferation of
smooth muscle in the lamina propria.
Classification (Sydney)
Modified Sydney’s
system, which was accepted in 1994 in Houston
1) Atrophic gastritis;
2) Non-atrophic
gastritis;
3) Special or
distinctive forms of gastritis: chemical, radiative,
lymphocytic,
eosinophilic, isolated granulomatous gastritis and
others.
6. Chronic gastritis. Symptoms and
methods of diagnostics. Treatment.
Diagnosis
CHRONIC ATROPHIC (AUTOIMMUNE) GASTRITIS
CHRONIC ATROPHIC (AUTOIMMUNE) GASTRITIS
Gastroscopy. The
diagnosis of chronic gastritis can only be ascertained histologically.
Therefore, histological assessment of endoscopic biopsies is essential. Tissue
sampling from both the gastric antrum and corpus is essential to establish the
topography of gastritis and to identify atrophy and intestinal metaplasia.
Laboratory studies.
Decreased serum pepsinogen I levels and the ratio of pepsinogen I to pepsinogen
II in the serum can be used to assess gastric atrophy. The finding of low
pepsinogen I levels (< 20 ng/mL) has a sensitivity of approximately 96.2%
and a specificity of 97% for detection of fundus atrophy. Antiparietal and
anti-intrinsic factor antibodies may be present in the serum. Low serum
cobalamin (vitamin B-12) levels (<100 pg/mL).
Gastric juices
analysis reveals achlorhydria, both basal and stimulated, and hypergastrinemia.
CHRONIC NON-ATROPHYC
microvascular architecture, as well as the mucosal surface
microstructure and tissue biopsy.
Methods of determining H. pylori:
Rapid urease test from gastric biopsy tissue.
Bacterial culture of gastric biopsy is usually performed in the
research setting or to assess antibiotic susceptibility in patients for
whom first-line eradication therapy fails.
Urea breath test with nonradioactive carbon isotope (13C) or
with radioactive carbon isotope (14C). The carbon 13 urea breath test
is based on the detection of the products created when urea is split by
the organism. Patients are asked to drink urea (usually with a
beverage) labeled with a carbon isotope (carbon 13 or carbon 14).
After a certain duration, the concentration of the labeled carbon is
measured in the breath. The concentration is high only when urease is
present in the stomach. Because the human stomach does not produce
urease, such a reaction is possible only with H. pylori infection.
H. pylori fecal antigen test. This is a novel rapid test based on
monoclonal antibody immunochromatography of stool samples. It has
been reported to be very specific (98%) and sensitive (94%). The
results are positive in the initial stages of infection and can be used to
detect eradication after treatment.
H. pylori serology. The serology test has a high (>90%)
specificity and sensitivity. It is currently based on the quantitation of
immunoglobulin G antibodies against H. pylori by the means of an
enzyme-linked immunosorbent assay. It is useful for detecting a newly
infected patient, but it is not a good test for follow-up of treated
patients because the results do not indicate present infection with
H. pylori. The antibody titer may remain elevated for a long time after
H. pylori eradication. The number of false-positive results is agerelated
and increases with age.
Treatment.
CHRONIC NON-ATROPHYC
CHRONIC NON-ATROPHYC
(HELICOBACTER PYLORI-ASSOCIATED) GASTRITIS
Gastroscopy is helpful for analyzing the subepithelialmicrovascular architecture, as well as the mucosal surface
microstructure and tissue biopsy.
Methods of determining H. pylori:
Rapid urease test from gastric biopsy tissue.
Bacterial culture of gastric biopsy is usually performed in the
research setting or to assess antibiotic susceptibility in patients for
whom first-line eradication therapy fails.
Urea breath test with nonradioactive carbon isotope (13C) or
with radioactive carbon isotope (14C). The carbon 13 urea breath test
is based on the detection of the products created when urea is split by
the organism. Patients are asked to drink urea (usually with a
beverage) labeled with a carbon isotope (carbon 13 or carbon 14).
After a certain duration, the concentration of the labeled carbon is
measured in the breath. The concentration is high only when urease is
present in the stomach. Because the human stomach does not produce
urease, such a reaction is possible only with H. pylori infection.
H. pylori fecal antigen test. This is a novel rapid test based on
monoclonal antibody immunochromatography of stool samples. It has
been reported to be very specific (98%) and sensitive (94%). The
results are positive in the initial stages of infection and can be used to
detect eradication after treatment.
H. pylori serology. The serology test has a high (>90%)
specificity and sensitivity. It is currently based on the quantitation of
immunoglobulin G antibodies against H. pylori by the means of an
enzyme-linked immunosorbent assay. It is useful for detecting a newly
infected patient, but it is not a good test for follow-up of treated
patients because the results do not indicate present infection with
H. pylori. The antibody titer may remain elevated for a long time after
H. pylori eradication. The number of false-positive results is agerelated
and increases with age.
Treatment.
CHRONIC NON-ATROPHYC
(HELICOBACTER PYLORI-ASSOCIATED)
GASTRITIS
`` Twice-daily proton pump
inhibitors or ranitidine
(lansoprazole 30 mg, omeprazole
20 mg, or ranitidine 400 mg
orally twice daily);
Clarithromycin 500 mg orally
twice daily;
Amoxicillin 1000 mg or
metronidazole 500 mg orally twice
daily.
Quadruple therapies (with
indicated adult dosing):
Proton pump inhibitors
(lansoprazole 30 mg or omeprazole 20
mg) orally twice daily;
Tetracycline HCl 500 mg orally
4 times daily;
Bismuth subsalicylate 120 mg
orally 4 times daily;
Metronidazole 500 mg orally 3
times daily.
CHRONIC ATROPHIC (AUTOIMMUNE) GASTRITIS
Once atrophic gastritis is
diagnosed, treatment can be directed to
correct complications of the
disease, especially in patients with
autoimmune atrophic gastritis
who develop pernicious anemia (in
whom vitamin B-12 replacement
therapy is indicated).
CHEMICAL GASTRITIS
Proton pump inhibitors have
demonstrated efficacy in controlled
trials for the treatment
chemical gastritis. An empiric 2-4 week trial of
an oral proton pump inhibitor
(omeprazole, rabeprazole, or
esomeprazole 20-40 mg/d;
lansoprazole, 30 mg/d; pantoprazole,
40 mg/d) is recommended.
7. Gastroduodenal ulcers. Etiology
and pathogenesis.
Etiology
The 2 major aetiologic factors
responsible for peptic ulceration are infection by the gram-negative gastric
pathogen Helicobacter pylori and the use of aspirin and other non-steroidal
anti-inflammatory drugs (NSAIDs). There is some synergy between these 2 major
causes. Duodenal ulcers are almost always
associated with H pylori infection, and gastric ulcers with NSAID use.
Rarer causes include gastric
ischaemia (responsible for the 'stress ulcers' that occur commonly in patients
with multiple organ failure in intensive care units), Zollinger-Ellison syndrome
(a syndrome of gastric acid hypersecretion caused by a gastrin secreting
neuro-endocrine tumour), certain medications (e.g., potassium chloride,
bisphosphonates), infections (CMV in patients with HIV, and occasionally HSV),
and Crohn's disease. A small but increasing proportion of peptic ulcers seem
truly idiopathic.
Pathogenesis
The normal stomach and
duodenum maintain a balance between the protective factors (mucus layer,
bicarbonate secretion, protective prostaglandins, blood flow) and aggressive
factors (gastric acid and proteolytic enzyme pepsin). Peptic ulcer disease
develops when aggressive factors overcome the protective mechanism. Any process
that increases gastric acidity (individuals with increased maximal and basal
acid output), decreases prostaglandin production (nonsteroidal
anti-inflammatory drugs, or interferes with the mucous layer (H. pylori
infection) can cause such an imbalance and leads to peptic ulcer disease.
8. Clinical manifestations of
gastroduodenal ulcers (ulcers of stomach and duodenal ulcer). Diagnostics
methods. Complications.
Clinical
picture
Complaints.
Patients may present with a wide variety of symptoms, or they may remain
completely asymptomatic. Classic gastric ulcer pain is described as pain
occurring shortly (15-20 min) after meals. The pain from gastric ulcer is
typically located in the epigastrium; however, it can also be perceived in the
right upper quadrant. Patients with gastric ulcer have vomiting, which relieve
stomach pain. So, in some cases patients provoke vomiting by
their selves to
relieve pain. Dyspepsia, including belching, bloating, distention, fatty food
intolerance may be present in gastric ulcer. Anorexia may occur with gastric
ulcers.
The epigastric pain in duodenal ulcer can be
sharp, dull, burning, or penetrating. It generally occurs 2-3 hours after meals
and often awakens the patient at night. Pain is often relieved by food. Many patients
experience a feeling of hunger. About 20-40% of patients describe bloating,
belching, or symptoms
suggestive of gastroesophageal reflux
(heartburn esophageal chest pain, regurgitation of acidic fluid).
Diagnostics methods
Upper
gastrointestinal radiography.
Endoscopy
Gastric juices
analysis
Complications.
Complications of
peptic ulcer disease include hemorrhage,
perforation,
penetration and gastric outlet obstruction. Patients with
gastric ulcers are
also at risk of developing gastric malignancy.
9. Treatment of gastroduodenal
ulcers.
Dietary factors
Diet may be of some
importance. Some products, which
stimulate gastric
acid secretion (coffee, alcohol), should be restricted
in acute cases.
Ulcer patients who smoke should be urged to decrease
or stop smoking.
Acid Neutralizing /
Inhibitory Drugs
Proton pump
inhibitors (omeprazole, esomeprazole,
lansoprazole,
rabeprazole, and pantoprazole) are the most potent acid
inhibitory agents
available.
H2-receptor
antagonists (ranitidine, famotidine, and nizatidine)
significantly
inhibit basal and stimulated acid secretion to comparable
levels when used at
therapeutic doses.
Antacids are now
rarely, if ever, used as the primary therapeutic
agent but instead
are often used by patients for symptomatic relief of
dyspepsia. The most
commonly used agents are mixtures of aluminum
hydroxide and
magnesium hydroxide (Maalox, Gaviscon).
Cytoprotective
Agents
Sucralfate is
insoluble in water and becomes a viscous paste
within the stomach
and duodenum, coats the ulcer bed and promotes
healing.
Bismuth induces
ulcer healing by ulcer coating; prevention of
further
pepsin/HCl-induced damage; binding of pepsin; and
stimulation of
prostaglandins, bicarbonate, and mucous secretion. but
does not decrease
gastric acid production. It is commonly used as one
of the agents in an
anti-H. pylori regimen.
Prostaglandin
analogues (Misoprostol) enhance mucous
bicarbonate
secretion, stimulate mucosal blood flow, and decrease
mucosal cell
turnover.
Therapy of H.
Pylori
H. pylori should be
eradicated in patients with documented
peptic ulcer
disease. H. pylori therapy is described previously (see
chronic
non-atrophic gastritis).
Surgery
Surgical
intervention in peptic ulcer disease can be viewed as
being either
elective, for treatment of medically refractory disease, or
as urgent/emergent,
for the treatment of an ulcer-related complication.
The development of
pharmacologic and endoscopic approaches for the
treatment of peptic
disease and its complications has led to a
substantial
decrease in the number of operations needed for this
disorder.
Refractory ulcers are an exceedingly rare occurrence.
Surgery is more
often required for treatment of an ulcer-related
complication.
gestive of gastroesophageal
reflux (heartburn esophageal chest pain, regurgitation of acidic fluid).
10. Special forms of ulcer localization: pyloric channel ulcers, multiple ulcers, giant ulcers. Stress ulcers.
11. Functional bowel disease.
Definition. Irritable bowel syndrome. Classification. Diagnostic criteria.
Irritable bowel syndrome (IBS) is a
functional gastrointestinal
disorder characterized by abdominal
pain and altered bowel habits in the absence of specific and unique organic
pathology.
Classification
IBS can be classified
as either diarrhea-predominant (IBS-D), constipation-predominant (IBS-C), or
with alternating stool pattern (IBS-A) or pain-predominant.
Clinical
picture
Complaints include abdominal pain, a feeling of incomplete
evacuation
(tenesmus), altered bowel habits, abdominal distention,
mucorrhea.
Abdominal pain. Pain frequently is intermittent, crampy without
radiation.
Acute episodes of sharp pain alternate on a more constant
dull
ache. Common sites of pain include the lower abdomen,
specifically
the left lower quadrant. The onset of pain typically is
associated
with a change in stool frequency or form and commonly is
relieved
by defecation.
Altered bowel
habits. Constipation variably results in complaints
of
hard stools of narrow caliber, painful or infrequent defecation, and
intractability
to laxatives. Diarrhea usually is described as small
volumes
of loose stool, with evacuation preceded by urgency or
frequent
defecation. Postprandial urgency is common. Alternating
habits
are common. Characteristically, one feature predominates in a
single
patient, but significant variability exists among patients.
Abdominal
distention. Patients frequently report increased
amounts
of bloating and gas. People with irritable bowel syndrome
may
manifest increasing abdominal circumference throughout the day.
Clear
or white mucorrhea of a noninflammatory etiology is
commonly
reported.
Diagnosis
clear
diagnostic markers exist for irritable bowel syndrome,
thus
the diagnosis of the disorder is based on clinical presentation. The
Rome
III criteria for the diagnosis of irritable bowel syndrome require
that
patients have had recurrent abdominal pain or discomfort at least
3
days per month during the previous 3 months that is associated with
2
or more of the following:
Abdominal pain
or discomfort characterized by the following:
-
Relieved by defecation
-
Associated with a change in stool frequency
-
Associated with a change in stool consistency.
Supporting
symptoms include the following:
-
Altered stool frequency
-
Altered stool form
-
Altered stool passage
-
Mucorrhea
-
Abdominal bloating or subjective distention.
Symptoms
not consistent with irritable bowel syndrome should
alert
the clinician to the possibility of an organic pathology.
Associated
symptoms and signs which increase the likelihood of
organic
disease include: recent onset (irritable bowel syndrome is
defined
by chronicity), progressive symptoms, blood in stools,
nocturnal
diarrhea, painless diarrhea, steatorrhea, weight loss, fever,
positive
HIV status. The absence of the above supports the diagnosis
of
irritable bowel syndrome.
Investigations
Blood test is normal.
Stool test will usually be negative for occult blood.
Colonoscopy
and barium enema are normal.
12. Treatment of irritable bowel
syndrome.
Therapy
Diet. Fiber supplementation may improve symptoms of
constipation and diarrhea. Individualize the treatment because few
patients experience exacerbated bloating and distention with highfiber
diets.
Pharmacologic treatment. The selection of pharmacologic
treatment remains symptom directed.
Anticholinergics (Dicyclomine hydrochloride (Bentyl),
Hyoscyamine sulfate (Levsin)) - are antispasmodics that inhibit
intestinal smooth-muscle depolarization at the muscarinic receptor.
Decrease fecal urgency and pain. Useful with diarrhea-predominant
symptoms.
Antidiarrheals (Loperamide (Imodium), Diphenoxylate
hydrochloride 2.5 mg with atropine sulfate 0.025 mg (Lomotil)) - are
nonabsorbable synthetic opioids. They prolong gastrointestinal tract
transit time and decrease secretion via peripheral _-opioid receptors.
Improve stool frequency and consistency, reduce abdominal pain and
fecal urgency, and may exacerbate constipation.
Prokinetics (metoclopramide, domperidone, Cisapride
monohydrate (Propulsid), Tegaserod (Zelnorm)) - are promotility
agents, proposed for use with constipation-predominant symptoms.
Accelerate gastrointestinal tract transit, thus increasing stool
frequency and improving consistency.
Tricyclic antidepressants (imipramine (Tofranil), amitriptyline) -
provide antidepressive and analgesic properties.
Bulk-forming laxatives (Methylcellulose (Citrucel), Psyllium
(Metamucil, Fiberall, Reguloid, Konsyl) - may use these agents as
fiber supplementation to improve symptoms of constipation and
diarrhea. Use is controversial.
13. Malabsorption syndrome.
Etiology. Pathogenesis. Clinical picture. Diagnosis. Therapy. Tropical
diarrhea.
Etiology and Pathogenesis
Malabsorption constitutes the pathological
interference
with the normal physiological sequence of digestion
(luminal phase), absorption (mucosal phase) and transport (postabsorptive
phase) of nutrients.
Luminal phase. Impaired nutrient hydrolysis (1)
pancreatic insufficiency), (2) inadequate mixing of nu trients due to rapid intestinal
transit (gastrojejunostomy, total and partial gastrectomy, intestinal
resection). Impaired fat solubilization (1) decreased bile salt synthesis from
severe parenchymatous liver disease (cirrhosis);(2) impaired bile secretion
from biliary obstruction or cholestatic jaundice (primary biliary cirrhosis,
primary sclerosing cholangitis); (3) impaired enterohepatic bile circulation,
as seen in small bowel resection or regional enteritis; (4) bile salt deconjugation
due to small bowel bacterial overgrowth. Decreased nutrient availability (1) cofactor
deficiency (gastric surgery), (2) bacterial overgrowth can cause a decrease in
the availability of substrates, including carbohydrates, proteins, and vitamins
(vitamin B12, folate).
Mucosal phase. Impaired nutrient absorption (1)
decreased
absorptive surface area, as seen in intestinal
resection of intestinal bypass; (2) damaged absorbing surface, as seen in
Celiac Sprue, Tropical Sprue, Crohn’s's disease, AIDS enteropathy,
chemotherapy, or radiation therapy; (3) infiltrating disease of the intestinal
wall, such as lymphoma and amyloidosis; and (4) infections, including bacterial
overgrowth, giardiasis, Whipple's disease, cryptosporidiosis, and microsporidiosis.
Malabsorption of specific substances (lactose intolerance).
Postabsorptive phase. Disturbances of mesenteric
circulation.
Obstruction of the lymphatic system, both
congenital (intestinal
lymphangiectasia, Milroy disease) and acquired (Whipple
disease, neoplasm [including lymphoma], tuberculosis), impairs the absorption of
chylomicrons and lipoproteins and may cause fat malabsorption or a
protein-losing enteropathy.
Clinical
picture
Steatorrhoea
Undigested
food in the stool
Weight loss and nutritional disturbances
Diarrhea
Flatulence and abdominal
distention
Edema
Muscle wasting and atrophy
Anemia
Dermatologic manifestations.
Neurologic manifestations
Diagnosis
Blood test: may reveal a macrocytic or microcytic anemia.
Biochemical blood test:may reveal low serum
levels of
cholesterol,
protein and albumin, sodium, potassium, chloride and
calcium, iron, vitamin B12, increased prothrombin time.
Stool fat analysis
Testing for unabsorbed carbohydrate. Unabsorbed
carbohydrate
produces stool with an acid pH of feces.
The urinary D-xylose test for carbohydrate
absorption provides
an assessment of proximal small-intestinal mucosal function.
The bile acid breath test is used if one
suspects bile acid
malabsorption
Barium contrast x-rays of the small intestine.
Endoscopic biopsy
Ultrasound, CT and MRI
Therapy
Dietary
modification is important in some conditions: glutenfree
diet in
celiac disease, lactose avoidance in lactose intolerance.
Replacement
therapy (replacement of nutrients, electrolytes and
fluid) is
used in patients with malabsorption syndromes.
Pancreatic enzymes
are supplemented orally in insufficiencies.
Enteral supplements,
bile salt binding agents, vitamins and minerals are also used in treatment.
Antibiotic
therapy will treat Small Bowel Bacterial overgrowth.
Cholestyramine
or other bile acid sequestrants will help reducing
diarrhea in
bile acid malabsorption.
Jejunostomy
is a surgical procedure done to allow placement of
feeding tube
as one treatment option for a variety of causes of
malabsorption.
Notion
of tropical diarrhea
Malabsorption due to small intestinal disease in a patient in or from
the tropics. There has to be an absence of other intestinal disease or
parasites. Its manifestations resemble those of celiac disease
Diarrhoea
Steatorrhoea or fatty stool (often foul-smelling and whitish in colour)
Indigestion
Cramps
Weight loss and malnutrition
Fatigue
Left untreated, nutrient and vitamin deficiencies may develop in
patients with tropical sprue.[1] These deficiencies may have the following
symptoms:
Vitamin A deficiency: hyperkeratosis or skin scales
Vitamin B12 and folic acid deficiencies: anaemia
Vitamin D and calcium deficiencies: spasm, bone pain, numbness and
tingling sensation
Vitamin K deficiency: bruises
Diagnosis
Abnormal flattening of villi and inflammation of the lining of the small
intestine, observed during an endoscopic procedure.
Presence of inflammatory cells (most often lymphocytes) in the biopsy of
small intestine tissue.
Low levels of vitamins A, B12, E, D, and K, as well as serum albumin,
calcium, and folate, revealed by a blood test.
Excess fat in the feces (steatorrhoea).
Thickened small bowel folds seen on imaging
The small bowel histological changes closely resemble Celiac disease,
although partial villous atrophy rather than subtotal villous is the usual lesion.
Treatment
Dehydration and electrolyte deficiencies must be
corrected in severe diarrhea
Tetracycline 1 g daily in divided doses for 28 days
(up to 6 months)
Folic acid and Vitamin B12 supplementation are given
as this relieves folate deficiency and improves
absorption
The small bowel mucosa soon returns to normal
14. Celiac disease. Etiology.
Pathogenesis. Clinical picture. Diagnosis. Principles of treatment.
Etiology
Autoimmune
Pathogenesis
Loss of immune tolerance to
gliadin peptide antigens derived from wheat, rye, barley, and related grains is
the central abnormality of coeliac disease. These peptides are resistant to
human proteases, allowing them to persist intact in the small intestinal lumen.
It is unknown how these peptides gain access to the lamina propria, but leading
hypotheses include faulty tight junctions, endothelial cell transcytosis,
sampling of the intestinal lumen by dendritic cells, and passage during
resorption of apoptotic villous enterocytes.
In the intestinal submucosa these
peptides trigger both innate and adaptive immune activation
Clinical
picture
The symptoms range
from significant malabsorption of multiple nutrients with diarrhea,
steatorrhea, weight loss, and the consequences of nutrient depletion (i.e.,
anemia and metabolic bone disease) to the absence of any gastrointestinal
symptoms but with evidence of the depletion of a single nutrient.
Diagnosis
blood antibody tests
The diagnosis of celiac sprue requires the presence of characteristic
histopathologic changes on small-intestinal biopsy together with a prompt
clinical and histopathologic response following the institution of a
gluten-free diet.
Therapy
About 90% of patients who have the characteristic findings of celiac
sprue will respond to complete dietary gluten restriction. n The remainder constitute a heterogeneous group
(whose condition is often called refractory sprue) that includes some patients
who (1) respond to restriction of other dietary protein, e.g., soy; (2) respond
to glucocorticoids; (3) are “temporary,” i.e., the clinical and morphologic
findings disappear after several months or years; or (4) fail to respond to all
measures and have a fatal outcome
15. Inflammatory bowel disease:
ulcerative colitis. Pathogenesis. Clinical features.

Etiology
The exact
etiology of ulcerative colitis is unknown, but
the disease
appears to be multifactorial and polygenic. Proposed
causes
include genetic factors, immune system reactions,
environmental
factors, non-steroidal anti-inflammatory drug use, low levels of antioxidants,
psychological stress factors.
Pathogenesis
1) Bacterial antigens are taken up by specialised M
cells, pass between
leaky epithelial cells or enter the lamina propria
through ulcerated mucosa.
(2) After processing, they are presented to type 1
T-helper cells by
antigen-presenting cells (APC) in the lamina
propria.
(3) T-cell activation
and differentiation results in a Th1 T
cell-mediated cytokine response
(4) with secretion of cytokines, including
interferon-gamma (IFN-γ). Further
amplification of T cells perpetuates the
inflammatory process with
activation of non-immune cells and release of other
important cytokines,
including interleukin (IL)-12, IL-23, IL-1 IL-6 and
tumour necrosis factor
(TNF). These pathways occur in all normal
individuals exposed to an
inflammatory insult and this is self-limiting in
healthy subjects. In
genetically predisposed persons, dysregulation of
innate immunity may
trigger inflammatory bowel disease.
Clinical
picture
depending on the severity stage
depending on the severity stage
Complaints. The main
complaints are diarrhea, rectal bleeding,
tenesmus,
passage of mucus, and different degrees of abdominal pain,
from
mild discomfort to painful bowel movements or painful
abdominal
cramping with bowel movements. Patients are commonly
fatigued,
which is often related to the inflammation and anemia that
accompany
disease activity. Weight loss is also common.
Variability
of symptoms reflects differences in the extent of
disease
(the amount of the colon and rectum that are inflamed) and the
intensity
of inflammation.
Ulcerative proctitis refers
to inflammation that is limited to the
rectum.
In many patients with ulcerative proctitis, mild intermittent
rectal
bleeding may be the only symptom. Other patients with more
severe
rectal inflammation may, in addition, experience rectal pain,
urgency
(sudden feeling the need to defecate and a need to rush to the
bathroom
for fear of incontinence), and tenesmus (ineffective, painful
urge
to move one's bowels).
Proctosigmoiditis involves
inflammation of the rectum and the
sigmoid
colon (a short segment of the colon contiguous to the rectum).
Symptoms
of proctosigmoiditis, like that of proctitis, include rectal
bleeding,
urgency, and tenesmus. Some patients with
proctosigmoiditis
also develop bloody diarrhea and cramps.
Left-sided colitis involves
inflammation that starts at the rectum
and
extends up the left colon (sigmoid colon and the descending
colon).
Symptoms of left-sided colitis include bloody diarrhea,
abdominal
cramps, weight loss, and left-sided abdominal pain.
Pancolitis or universal colitis refers to inflammation affecting
the
entire colon (right colon, left colon, transverse colon and the
rectum).
Symptoms of pancolitis include bloody diarrhea, abdominal
pain
and cramps, weight loss, fatigue, fever, and night sweats. Some
patients
with pancolitis have low-grade inflammation and mild
symptoms
that respond readily to medications. Generally, however,
patients
with pancolitis suffer more severe disease and are more
difficult
to treat than those with more limited forms of ulcerative
colitis.
Fulminant colitis is
a rare but severe form of pancolitis. Patients
with
fulminant colitis are extremely ill with dehydration, severe
abdominal
pain, protracted diarrhea with bleeding, and even shock.
They
are at risk of developing toxic megacolon (marked dilatation of
the
colon due to severe inflammation) and colon rupture (perforation).
Patients
with fulminant colitis and toxic megacolon are treated in the
hospital
with potent intravenous medications. Unless they respond to
treatment
promptly, surgical removal of the diseased colon is
necessary
to prevent colon rupture.

16. Inflammatory bowel disease:
ulcerative colitis. Diagnosis and principles of treatment.
Diagnosis
Blood test reveals
anemia, thrombocytosis, and elevated ESR,
which
correlate with disease activity.
Biochemical blood test reveals
hypoalbuminemia, hypokalemia,
hypomagnesemia,
elevated alkaline phosphatase and elevated Creactive
protein.
Serologic test. Perinuclear
antineutrophil cytoplasmic antibody
(p-ANCA),
a myeloperoxidase antigen, is more commonly found in
ulcerative
colitis.
Stool test is used
to exclude infection and parasites, since these
conditions
can cause colitis that mimics ulcerative colitis.
Colonoscopy reveals
edema, hyperemia of the mucosa.
Ulcerations
and mucopurulent exudates may be present. Islands of
normal
tissue may have the appearance of pseudopolyps. Numerous
biopsy
samples should be obtained.
Barium contrast studies. The
earliest radiologic change of
ulcerative
colitis is a fine mucosal granularity. With increasing
severity,
the mucosa becomes thickened, and ulcers are seen. Haustral
folds
may be normal in mild disease, but as activity progresses they
become
edematous and thickened. In addition, the colon becomes
shortened
and narrowed. There may be postinflammatory polyps or
pseudopolyps.
Therapy
The
goals of treatment with medication are to 1) induce
remissions,
2) maintain remissions, 3) minimize side effects of
treatment,
4) improve the quality of life, and 5) minimize risk of
cancer.
Medications
used in reducing inflammation in Ulcerative colitis
include
anti-inflammatory drugs, corticosteroids, immunosuppressant
agents,
antimicrobials and antidiarrheal medicines.
Anti-inflammatory drugs.
These agents have anti-inflammatory
effects.
They are used to maintain remission. Sulfasalazine
is useful
in
treating mild-to-moderate ulcerative colitis and maintaining
remission.
It acts locally in the colon to reduce the inflammatory
response
and systemically inhibits prostaglandin synthesis.
Mesalamine is the
drug of choice for maintaining remission. It is
useful
for the treatment of mild-to-moderate ulcerative colitis. It is
better
tolerated and has less adverse effects than sulfasalazine. Enema
and
suppository forms are typically used in patients with distal colitis.
Tumor Necrosis Factor Inhibitor. These agents prevent the
endogenous
cytokine from binding to cell surface receptor and
exerting
biological activity. These agents adversely affect normal
immune
responses. Infliximab is a chimeric mouse-human
monoclonal
antibody to tumor necrosis factor alpha. Infliximab is
indicated
for the treatment of moderate-to-severe active ulcerative
colitis
in patients who have experienced inadequate response to
conventional
therapy. Adalimumab is a recombinant human antitumor
necrosis
factor alpha IgG1 monoclonal antibody that blocks
inflammatory
activity of TNF-alpha. This agent is indicated for
ulcerative
colitis unresponsive to immunosuppressant medicines
(corticosteroids,
azathioprine, 6-mercaptopurine).
Immunosuppressant Agent.
These agents regulate key factors of
the
immune system. Azathioprine (Imuran) is effective as a steroidsparing
or
steroid-reducing agent and for use in maintenance therapy.
Administration
is oral. Onset of action can be delayed up to 3-6
months.
Cyclosporine is effective as a means of avoiding surgery in
patients
with severe ulcerative colitis refractory to intravenous
corticosteroids.
It is given as an intravenous infusion. 6-
Mercaptopurine is
effective as a steroid-reducing or steroid-sparing
agent
and for use in maintaining remission. Administration is oral.
Onset
of action can be delayed up to 3-6 months.
Antimicrobials. In
several controlled, trials, antibiotics have not
been
shown to provide consistent benefits for the treatment of active
ulcerative
colitis. Thus, they are usually administered on an empiric
basis
in patients with severe colitis in whom they may help with
averting
a life-threatening infection. Ciprofloxacin,
Metronidazole
can
be used.
Corticosteroids. Corticosteroids
decrease inflammation by
suppressing
migration of polymorphonuclear leukocytes and reversing
increased
capillary permeability. They are used for induction of
remission
in moderate-to-severe active ulcerative colitis. They have
no
benefit in maintaining remission; long-term use can cause adverse
effects.
Methylprednisolone, Prednisone,
Hydrocortisone can be
used
in treatment of Ulcerative Colitis.
Antidiarrheal. These
agents are nonabsorbable synthetic
opioids
that provide symptomatic relief in the treatment of ulcerative
colitis.
They prolong gastrointestinal transit time and decrease
secretion.
Loperamide (Imodium) improves stool frequency and
consistency, reduces abdominal pain and fecal urgency.
17. Definition of chronic colitis
(non-ulcer). Etiology. Pathogenesis. Clinical features. Factors associated with
the development of chronic colitis.
18. Chronic colitis (non-ulcer).
Diagnosis and differential diagnosis. Treatment.
19. Inflammatory bowel disease:
Crohn's disease. Pathogenesis. Clinical features. Diagnosis and principles of
treatment.
Etiology
Etiology
The exact cause of Crohn’s disease remains unknown. Current
theories implicate the role of genetic, microbial, immunologic, environmental, dietary, vascular, and even psychosocial
factors as potential causative agents. It has been suggested that patients have an inherited susceptibility for an aberrant
immunologic response to one or more of these provoking factors.
Pathogenesis
1) Bacterial antigens are taken up by specialised M cells, pass between
leaky epithelial cells or enter the lamina propria through ulcerated mucosa.
(2) After processing, they are presented to type 1 T-helper cells by
antigen-presenting cells (APC) in the lamina propria.
(3) T-cell activation
and differentiation results in a Th1 T cell-mediated cytokine response
(4) with secretion of cytokines, including interferon-gamma (IFN-γ). Further
amplification of T cells perpetuates the inflammatory process with
activation of non-immune cells and release of other important cytokines,
including interleukin (IL)-12, IL-23, IL-1 IL-6 and tumour necrosis factor
(TNF). These pathways occur in all normal individuals exposed to an
inflammatory insult and this is self-limiting in healthy subjects. In
genetically predisposed persons, dysregulation of innate immunity may
trigger inflammatory bowel disease.
Clinical picture depending on the severity stage
Clinical features are characterized by periodic exacerbations and
remissions.
Complaints. The main complaints are abdominal pain, diarrhea,
meteorism (flatus) and bloating, low-grade fever, weight loss and
generalized fatigue.
Pain is often crampy or steady in the right lower quadrant or
periumbilical area. It may be colicky, especially in the lower
abdomen.
Diarrhea is nonbloody and often intermittent. If the colon is
involved, patients may report diffuse abdominal pain accompanied by
mucous, blood and pus in the stool.
Patients may also present with complaints suggestive of
intestinal obstruction: postprandial bloating, cramping pain increased
after meals and relieved by defecation, and loud borborygmi. Once the
bowel lumen becomes chronically narrowed, patients complain of
constipation and obstipation.
Therapy
The goals of treatment of Crohn's disease are to reduce the
underlying inflammation, which then relieves symptoms, prevents
complications, and maintains good nutrition.
Medications used in reducing inflammation in Crohn's disease
include anti-inflammatory drugs, corticosteroids, other
immunosuppressants and antibiotics.
Anti-inflammatory drugs. Mesalamine reduces the
inflammation, tends to work best in Crohn's disease affecting mainly the colon and to some extent the small intestine. Oral and rectal suppository forms are available. Long-term use may delay relapse of the disease. Sulfasalazine tends to work best in Crohn's disease affecting mainly the colon. It does not work well in the small intestine.Long-term use generally does not delay relapse.
Corticosteroids: Prednisone and Budesonide reduce
inflammation and suppress the immune system. They can be used in the short term only. Corticosteroids are indicated in persons with severe systemic symptoms (for example, fever, nausea, weight loss) and in those who do not respond to anti-inflammatory agents.
Immunosuppressants: Azathioprine, methotrexate, infliximab,
adalimumab, certolizumab and natalizumab may be used in treatment of Crohn's disease. Infliximab (Remicade) is a monoclonal antibody that acts against tumor necrosis factor alpha, a natural product of the immune system that promotes inflammation. Infliximab is used to treat moderately severe-to-severe Crohn's disease that does not get better with other medications. When given as an intravenous infusion, its effects last for approximately 12 weeks. Repeated doses may be required.
Antibiotics reduce inflammation indirectly by reducing infection.
Metronidazole, besides acting as an antibiotic, has
immunosuppressive and anti-inflammatory properties. Ciprofloxacin, Cephalosporin, Ampicillin, Tetracycline, Sulfonamide, and others can be used.
20. Inflammatory bowel disease:
ulcerative colitis and Crohn's disease. Differential diagnosis and
complications (intestinal and extraintestinal).
21. Chronic cholecystitis.
Etiology. Pathogenesis.
Etiology and
pathogenesis-
Chronic inflammation of the
gallbladder wall is almost always associated with the presence of gallstones
and is thought to result from repeated bouts of subacute or acute cholecystitis
or from persistent mechanical irritation of the gallbladder wall by gallstones.
Role of infection in
cholecystitis-
The presence of bacteria in the
bile occurs in >25% of patients with chronic cholecystitis.
The presence of infected bile in
a patient with chronic cholecystitis undergoing elective cholecystectomy
probably adds little to the operative risk. Chronic cholecystitis may be
asymptomatic for years, may progress to symptomatic gallbladder diseasesor to
acute cholecystitis, or may present with complications.
Cholelithiasis-
Choledocholithiasis (stones in
common bile duct) is one of the complications of cholelithiasis (gallstones).
This condition causes mechanical jaundice and liver cell damage, and requires
treatment by cholecystectomy.
Patients with cholelithiasis typically present
with pain in the right-upper quadrant of the abdomen with the associated
symptoms of nausea and vomiting, especially after a fatty meal.
Patients may have jaundice, dark
urine and pale feces.
Physical examination. Jaundice
of the skin or eyes is an important physical finding in biliary obstruction.
Murphy's sign is often negative
on physical examination in choledocholithiasis, helping to distinguish it from
cholecystitis.
In this case Biochemical blood test may reveal
elevation in alkaline phosphatase, increase in conjugated bilirubin and
increase cholesterol level.
The diagnosis is confirmed with either a magnetic resonance
cholangiopancreatography, an endoscopic retrograde cholangiopancreatography
(ERCP) or an intraoperative cholangiogram.
Treatment involves removing the stone using
ERCP. Typically, the gallbladder is then removed, an operation called
cholecystectomy, to prevent a future occurrence of common bile duct obstruction
or other complications.
22. Chronic cholecystitis. The
major symptoms and diagnosis of chronic cholecystitis. Complications.
Principles of treatment.
Clinical
picture-
Chronic
cholecystitis is asymptomatic.it may progress with symptomatic gall bladder
disease or acute cholecystitits or may present with it’s complications .
Diagnosis
in strumental and laoratory diagnosis-
Blood test. -
Leucocytes count
results are normal in uncomplicated biliary colic; an abnormality suggests
complicated biliary disease such as cholecystitis.
Biochemical blood
test-
AST, ALT, alkaline
phosphatase, bilirubin, and amylase assay results are normal in uncomplicated
biliary colic; an abnormality suggests a complication such as cholecystitis,
cholangitis, or pancreatitis.
Ultrasonography-
Ultrasonography is
the most sensitive and specific test for the detection of gallstones.
It provides
information about the size of the common bile duct and hepatic duct and the
status of liver parenchyma and the pancreas.
Thickening of the
gallbladder wall and the presence of pericholecystic fluid are radiographic
signs of acute cholecystitis.
Complication
-
Passage of gallstones from the gallbladder into the common bile
duct can result in a complete or partial obstruction of the common bile duct.
Frequently, this
manifests as mechanical/obstructive jaundice.
Pancreatitis,
another complication of gallstone disease, presents with more diffuse abdominal
pain, including pain in the epigastrium and left upper quadrant of the abdomen.
Charcot triad (right
upper quadrant pain, fever, and jaundice) is associated with common bile duct
obstruction and cholangitis.
GanGrene anD
perforation Gangrene of the gallbladder results from ischemia of the wall and
patchy or complete tissue necrosis.
fistula formation
and Gallstone ileus Fistula formation into an adjacent organ adherent to the
gallbladder wall may result from inflammation and adhesion formation. Fistulas
into the duodenum are most common, followed in frequency by those involving the
hepatic flexure of the colon, stomach or jejunum, abdominal wall, and renal
pelvis.
limey (milk of
calcium) bile and porcelain Gallbladder - Calcium salts in the lumen of the
gallbladder in sufficient concentration may produce calcium precipitation and
diffuse, hazy opacification of bile or a layering effect on plain abdominal
roentgenography.
Therapy-
-Medical
Cholesterol
gallstones can sometimes be dissolved by oral ursodeoxycholic acid, but it may
be necessary for the patient to take this medication for up to two years.
Gallstones may recur, however, once the drug is stopped.
Obstruction of the common
bile duct with gallstones can sometimes be relieved by endoscopic retrograde
sphincterotomy following endoscopic retrograde cholangiopancreatography.
Gallstones can be
broken up using a procedure called extracorporeal shock wave lithotripsy (often
simply called "lithotripsy"), which is a method of concentrating
ultrasonic shock waves onto the stones to break them into tiny pieces.
They are then passed
safely in the feces. However, this form of treatment is suitable only when
there is small number of gallstones.
-Surgical.
Cholecystectomy (gallbladder removal) has a 99% chance of eliminating the
recurrence of cholelithiasis. Surgery is only indicated in symptomatic
patients.
The lack of a
gallbladder may have no negative consequences in many people.
However, there is a
portion of the population - between 10 and 15% - who develop a condition called
postcholecystectomy syndrome which may cause gastrointestinal distress and
persistent pain in the upper-right abdomen, as well as a 10% risk of developing
chronic diarrhea.
Diet-
Avoid fried foods ,fatty foods and particular
fats like hydrogenated fats,saturated fats.
Avoid alcohol
intake, reduce the ammount od carbohydrates.
Take more green
vegetables and fruits.
Indication
for surgery-
Common bile duct
obstruction
Asymptomatic gall
stones
One or more episodes
of biliary pain
Previous diagnose of
acute cholecystitis.
23. Chronic pancreatitis.
Definition. Pathogenesis. Classification.
Definition-
Chronic pancreatitis
is characterised by chronic inflammation, calcification and atrophy of the
pancreas. It is caused by alcohol in >75% of cases.
The early disease is
dominated by chronic abdominal pain, with time pain diminishes and exocrine and
endocrine insufficiency dominate.
Etiologic
risk factors-
Alcohol and smoking
Biliary tract
disease
Iatrogenic
Metabolic disorders
,hyper glyceridaemia,hypercalcaemia.
Trauma of pancreas
Congenital disorders
eg-cystic fibrosis
Auto immune
pancreastitis
Drug eg-sulfonamides,loop
diuretics.
Idiopathic-thought
to be early and late onset subgroups.
Abdominal
radiotherapy-causes of obstruction CP include pancreatic
adenocarcinoma,neuroendocrine tumors and intra papillary mucinous tumors.
Haemochromatosis-
genetic disease caused by too much iron in the body.
Pancreas
divisum-patient born with duct which do not function properly.
Pathogenesis-
Pathogenic theories
have been developed including
1-oxidative stress
2-toxic metabolic
3-stone with duct
obstruction
4-necrosis -fibrosis
Oxidative stress-
Products of hepatic
mixed function oxidase activity -> chronic reflux of bile into pancreatic
duct -> damage of pancreas
Toxic metaboilc-
Alcohol changing the
intracellular metabolism->direct toxic to aciner cells ->produce
pancreatic lipid accumilation,fattty degeneration,cellular necrosis ->
widespread fibrosis
Stone with duct
obstruction-
Alcohol intake
increase lithogenicity of pancreatic juice -> stone formation ->chronic
contact of stones with epithelial cells ->ulceration and scarformation
->atrophy and fibrosis ->obstruction of the acne.
Necrosis fibrosis
theory-
Scarring and
extrinsic compression of the pancreatic ductules ->obstruction ->
stasis,atrophy,stone formation.
Classification-
1)by etiological sign-
A-primary (initial) pancreatitis- inflammatory process in the pancreas only
B- secondary pancreatits- disease spread in
other organs of GIT, this is long term period.
2)bu morphological sign-
Edematous form
Sclerotic form
Fibrous
Pseudocytic
With calcification
of the pancreas
3)by special features of the clinical pictures-
A-chronic recurrency(relapsing) pancreatits-
During the period
between the attack the patient feels satisfactory.
B-chronic painful pancreatitis-
C-chronic unpainful pancreatitis-
D-chronic pseudocytic-cyst which compresses
the common bile duct,duodenum ,pylorus.
E-chronic cholecysto pncreatitis-with
symptoms of chronic cholecystitis and chronic pancreatitis.
F-chronic calculous cholecystitis-calicum
deposit in ducts and parenchyma of pancreas.
4)by the cause of the disease-
A- mild degree of severity
B-a moderate degree of severity
C- a severe forrm
24. Chronic pancreatitis. Symptoms
and methods of diagnostics. Treatment.
Instrumental
and laboratory methods
of examination (ultrasound, X-ray, CT).
of examination (ultrasound, X-ray, CT).
Clinical
picture-
Abdominal Pain -pain
may begin gradually and persist for days or weeks.
Pain is located in
the epigastrium, right or left subcostal areas or around the umbilicus,
characteristically it may radiate to the back and relief may be obtained by crouching
forward or leaning forward over a chair.
The patient may also
complain about pain related to their food intake, especially those meals
containing a high percentage of fats and protein.
Pain with nausea
vomiting.
Decrease appetite
exocrine insufficiency
due to this malabsorption
,diarrhea,weight loss steatorrhea with foul smell and greasy stool.
Diabetes is a common
complication due to the chronic pancreatic damage and may require treatment
with insulin.
During physical
examination a tender fullness or mass may be palpated in the epigastrium,
suggesting the presence of a pseudocyst or an inflammatory mass in the
abdomen.
A few patients with
severe, intractable pain exhibit a characteristic mottled erythema or
hyperpigmentation over their abdomens or back; on further questioning, these
patients will reveal that they sustained burns to the skin while applying hot
water bottles, heating pads, or electric pads over the area in an attempt to
relieve pain.
Instrumental
and laboratory methods of diagnosis-
Laboratory-
-Biochemical blood
test. Elevations of serum amylase and lipase are found only during acute
attacks of pancreatitis, usually early in the course of the disease.
In the later stages
of chronic pancreatitis, atrophy of the pancreatic parenchyma can result in
serum enzyme levels within the reference range, even during acute attacks of
pain.
-Serum bilirubin and
alkaline phosphatase can be elevated, indicating stricture of the common bile
duct due to edema, fibrosis or cancer.
-Feces analysis. The
common symptom of chronic pancreatitis, steatorrhea, can be diagnosed by two
different studies: Sudan chemical staining of feces or fecal fat excretion of 7
grams or more over a 24hr period on a 100g fat diet.
To check for
pancreatic exocrine dysfunction, the most sensitive and specific test is the
measurement of fecal elastase, which can be done with a single stool sample,
and a value of less than 200 ug/g indicates pancreatic insufficiency.
Instrumental-
-Abdominal x-ray.
Pancreatic calcifications, often considered pathognomonic of chronic
pancreatitis, are observed in approximately 30% of cases. Computed tomography scan.
This study is
indicated to look for complications of the disease and is useful in planning
surgical or endoscopic intervention.
-Endoscopic
retrograde cholangiopancreatography (ERCP) provides the most accurate
visualization of the pancreatic ductal system and has been regarded as the
criterion standard for diagnosing chronic pancreatitis.
-Magnetic resonance
cholangiopancreatography (MRCP) imaging provides information on the pancreatic
parenchyma and adjacent abdominal viscera, and MRCP uses heavily T2-weighted
images to visualize the biliary and pancreatic ductal system.
-Endoscopic
ultrasonography. Recent studies suggest that endoscopic ultrasonography may be
the best test for imaging the pancreas.
By placing the transducer immediately adjacent
to the pancreas, the endoscopic approach eliminates interference by bowel gas
and enables the use of high-frequency transducers to provide more detailed
imaging.
Eleven sonographic
criteria have been developed that identify characteristic findings of chronic
pancreatitis.
Plain x-ray of
abdomen
Angiography and
venography.
Barium swallow
Biopsy of pancreas.
Therapy-
The goals of
treatment are as follows:
Modify lifestyle
that may exacerbate the natural history of the disease Enable the pancreas to
heal itself Determine the cause of abdominal pain and alleviate it
Detect pancreatic
exocrine insufficiency and restore digestion and absorption to normal
Diagnose and treat endocrine insufficiency.
In early stage
alcohol-induced chronic pancreatitis, lasting pain relief can occur after
abstinence from alcohol, but in advanced stages, abstinence does not always
lead to symptomatic improvement.
Patients continuing
to abuse alcohol develop either marked physical impairment or have a death rate
3 times higher than do patients who abstain.
Analgesics.
The abdominal pain
can be very severe and require high doses of analgesics, sometimes including
opiates. Initial therapy consists of acetaminophen or nonsteroidal
anti-inflammatory drugs (NSAIDs).
For severe,
refractory pain, narcotic analgesics often are required, starting with the
least potent agents and progressing to more potent formulations as
necessary.
Pancreatic Enzyme
Supplementation. These are used as dietary supplementation to aid digestion in
patients with pancreatic enzyme deficiency.
Several preparations
are available: Pancrelipase (Creon, Pancreaze, Ultresa, Viokace, Zenpep).
The aim is to
provide at least 30,000 units of lipase. Surgery. Traditional surgery for
chronic pancreatitis tends to be divided into two areas - resection and
drainage procedures.
New and proven
transplantation options prevent the patient from becoming diabetic following
the surgical removal (resection) of their pancreas.
This is achieved by
transplanting back in the patient's own insulinproducing beta cells.
Diet-
Diet and life-style
changes. Cessation of alcohol consumption and tobacco smoking are important.
Eating low fat diet
is very important.
Include adequate fat
soluble vitamins and calcium in diet.
Medicamental-
Antidepressants.in
addition to alleviating coexistent depression, tricyclic antidepressants may
ameliorate pain and potentiate the effects of opiates.
Indications
for surgery-
1-If the pain become
sever and cannot treated by pain killers.
2-Pancreatic cancer
3-there are surgical
complications of chronic pancreatitis.
25. Chronic hepatitis. Etiology.
Classification (Los-Angeles). The major symptoms (describe the clinical
syndromes).
Etiology
Causes
of chronic hepatitis are: viruses, autoimmune
disorders,
alcohol, toxic influence, drugs induced, metabolic disorders
(amyloidosis,
fatty liver, disturbances in protein metabolism, copper
and
vitamins metabolism).
Viral Hepatitis. Viral
Hepatitis B and C are the most common
causes
of chronic hepatitis. Hepatitis B (HBV) is a DNA virus which
is
transmitted parenterally. Individuals at high risk include intravenous
drug
abusers, homosexual men and those exposed to blood and blood
products.
The incubation period ranges from 1 to 6 months. Chronic
HBV
develops in 10% of patients.
Hepatitis C (HCV) is
an RNA virus that formerly accounted for
90%
of post-transfusion hepatitis. The modes of transmission
(parenteral,
sexual and perinatal) are similar to those of HBV. The
incubation
period is 2 weeks to 6 months. Chronic hepatitis develops
in
up to 85% of patients. Cirrhosis may develop in 20% of patients,
with
an increased risk of hepatocellular carcinoma.
Hepatitis D (Delta
hepatitis, HDV) is caused by a small,
defective
RNA virus that is infectious only in presence of HBV
infection.
It can complicate acute HBV infection, but is seen more
commonly
as a superinfection in patient with chronic HBV. The
combination
of hepatitis B and D is worse than hepatitis B alone and
is
more likely to cause serious chronic hepatitis and cirrhosis.
Autoimmune Hepatitis. Autoimmune hepatitis is now accepted
as
a chronic disease of unknown cause, characterized by continuous
hepatocellular
inflammation and necrosis, which tends to progress to
cirrhosis.
The trigger for autoimmune chronic hepatitis is unknown,
but
the damage to the liver is caused by the individual's lymphocytes
and
by antibodies produced in the individual's own tissue. Most of the
patients
are young women but postmenopausal women and males may
get
the disease. Autoimmune chronic hepatitis is usually a progressive
disease
ending in cirrhosis.
Alcoholic Hepatitis.
Alcoholic hepatitis is a syndrome of
progressive
inflammatory liver injury associated with long-term heavy
intake
of ethanol. The pathogenesis is not completely understood.
Alcoholic
hepatitis usually persists and progresses to cirrhosis if
heavy
alcohol use continues.
Drug-Induced Hepatitis. Few medications still in use and
several
that have been withdrawn from the market can also cause
chronic
hepatitis. These include: isoniazid - used for tuberculosis;
methyldopa
- used for hypertension; nitrofurantoin - used for urinary
tract
infections; phenytoin - used for seizure disorders and some other
medications.
These medications must be taken for long periods of
time.
However, the number of cases of chronic hepatitis produced by
these
medications is small. Chronic hepatitis caused by drugs is
usually
recognized early. Stopping the medicine before cirrhosis has
developed
usually reverses the disease.
Inherited Disorders. Some
inherited disorders of metabolism
also
can appear as chronic hepatitis. The most frequent of these
conditions
is Wilson's disease - a familial disorder of copper
metabolism.
Alpha-1-antitrypsin deficiency and tyrosinemia may
appear
as chronic hepatitis although other features help in
distinguishing these rare conditions from those caused by
viruses.
Classification of
chronic hepatitis (Los Angeles, 1994)
1. Autoimmune
hepatitis.
2. Chronic hepatitis
B.
3. Chronic
hepatitis B and D
4. Chronic
hepatitis C.
5. Chronic
hepatitis (which is not characterized otherwise).
6. Chronic
hepatitis (not classified as viral or autoimmune).
7. Chronic
hepatitis drug.
Chronic hepatitis E
have been made to the classification, since the virus was discovered in 1995
after the approval of the classification.
26. Features of the pathogenesis of
viral hepatitis, autoimmune hepatitis.
viral hepatitis,
viral hepatitis,
Under ordinary circumstances, none of the
hepatitis viruses is known to be directly cytopathic to hepatocytes.
Evidence suggests that the clinical
manifestations and outcomes after acute liver injury associated with viral
hepatitis are determined by the immunologic responses of the host. Among the
viral hepatitides, the immunopathogenesis of hepatitis B and C has been studied
most extensively.
Hepatitis B (HBV)-
is a DNA virus which is transmitted
parenterally. Individuals at high risk include intravenous drug abusers,
homosexual men and those exposed to blood and blood products. The incubation
period ranges from 1 to 6 months. Chronic HBV develops in 10% of patients.
Hepatitis C -
The modes of transmission (parenteral, sexual
and perinatal) are similar to those of HBV. The incubation period is 2 weeks to
6 months.
HCV can cause direct liver injury or indirect
liver injury through immune‐mediated pathways. Pathways related to necrosis,
apoptosis, angiogenesis are all up‐regulated leading to progressive fibrosis.
Hepatitis D (Delta hepatitis, HDV) -
is caused by a small, defective RNA virus
that is infectious only in presence of HBV infection. It can complicate acute
HBV infection, but is seen more commonly as a superinfection in patient with
chronic HBV.
autoimmune hepatitis
IMMUNOPATHOGENESIS-
The weight of evidence suggests that the
progressive liver injury in patients with autoimmune hepatitis is the result of
a cell-mediated immunologic attack directed against liver cells.
In all likelihood, predisposition to
autoimmunity is inherited, whereas the liver specificity of this injury is
triggered by environmental (e.g., chemical, drug [e.g., minocycline], or viral)
factors.
Cellular immune mechanisms appear to be
important in the pathogenesis of autoimmune hepatitis. In vitro studies have
suggested that in patients with this disorder, CD4+ T lymphocytes are capable
of becoming sensitized to hepatocyte membrane proteins and of destroying liver
cells.
Molecular mimicry by cross-reacting antigens
that contain epitopes similar to liver antigens is postulated to activate these
T cells, which infiltrate, and result in injury to, the liver.
Humoral immune mechanisms have been shown to
play a role in the extrahepatic manifestations of autoimmune and idiopathic
hepatitis. Arthralgias, arthritis, cutaneous vasculitis, and glomerulonephritis
occurring in patients with autoimmune hepatitis appear to be mediated by the
deposition of circulating immune complexes in affected tissue vessels, followed
by complement activation, inflammation, and tissue injury.
27. Clinic of chronic hepatitis.
Diagnostics. Degrees of activity of chronic hepatitis.
28. Treatment of chronic viral
hepatitis. Treatment of autoimmune hepatitis.
Chronic viral hepatitis therapy-
The goal of treatment of the chronic
hepatitis B virus and chronic hepatitis C virus are to reduce the fibrosis and
inflammation and to prevent progression to cirrhosis and it’s complications.
Medications For hepatitis B virus includes
interferon (IFN) alpha-2a and the oral nucleoside or nucleotide analogues
example lamivudine and adefovirdipivoxil.
Medication for hepatitis C virus includes IFN
alfa-2b,IFN alfa-2a,consensus IFN( IFN alfacon-1,ribavirin in combination with
IFN.
29. Liver cirrhosis. Etiology.
Classification.
Etiology-
Cirrhosis is a diffuse hepatic process characterized by
fibrosis and the conversion of normal liver architecture into structurally
abnormal nodules, resulting in the loss of liver function.
The progression of liver injury to cirrhosis may occur
over weeks to years. Causes:
Alcoholic liver disease
Chronic viral hepatitis B, C and D
chronic autoimmune hepatitis
Inherited metabolic diseases (hemochromatosis,
Wilson disease, α1 Antitrypsin deficiency)
Biliary cirrhosis (primary biliary cirrhosis,
primary sclerosing cholangitis,
autoimmune
cholangiopathy,
secondary biliary cirrhosis - associated with chronic
extrahepatic bile duct obstruction)
Chronic congestive
heart failure (Cardiac cirrhosis) Parasitic infections (schistosomiasis)
Non-alcoholic steatohepatitis
Long term exposure
to toxins or drugs Cryptogenic cirrhosis (unknown cause).
Classification-
Classification of cirrhosis is based on
(1) its cause;
(2) size of the nodules;
(3) severity.
Depending on the size of the nodules there are three
macroscopic types of cirrhosis:
micronodular, macronodular and mixed.
In micronodular form (Laennec's cirrhosis or portal
cirrhosis) regenerating nodules are under 3 mm.
In macronodular cirrhosis (postnecrotic cirrhosis), the
nodules are larger than 3 mm.
The mixed cirrhosis consists in a variety of nodules with
different sizes.
The severity of cirrhosis is commonly classified with the
ChildPugh score.
This score uses bilirubin, albumin, INR, presence and
severity of ascites and encephalopathy to classify patients in class A, B or C.
Class A has a
favourable prognosis, while class C is at high risk of death. It was devised in
1964 by Child and Turcotte and modified in 1973 by Pugh.
30. Pathogenesis of liver
cirrhosis.
Liver cirrhosis cause by alcohol,hepatitis c virus
infection ,non alcoholic diseases , non alcoholic liver cirrhosis alpha anti
trypsin deficiency,hemachromatosis , wilson disease,primary
cirrhosis,sclerosing cholangitis.
Due to these diseases liver develop severe
inflammation,for an extended period of the time.
Over a long time fibrosis develop.
Development of scar tissue that replace normal parenchyma, blocking the portal flow
of blood through the organs.
When at least 70-80% of the liver function has been
lost,the synthetic capacity of the liver is diminished.
31. Primary biliary cirrhosis.
Etiology and pathogenesis. Clinical picture.
Etiology-
PBC is seen in about 100–200 individuals per million,
with a strong female preponderance and a median age of around 50 years at the
time of diagnosis.
The cause of PBC is unknown; it is characterized by
portal inflammation and necrosis of cholangiocytes in small- and medium-sized
bile ducts.
Cholestatic features prevail, and biliary cirrhosis is
characterized by an elevated bilirubin level and progressive liver failure.
Pathogenesis-
Bilirubin levels do not elevate untill the disease is
extremely far advanced,which is usually after 5 - 10 years.
PBC has a strong association with other auto immune
disease,such as sjogren syndrom (dry eyes and dry mouth )
Rheumatoid arthritis and scleroderma.
Clinical features-
most patients are actually asymptomatic. When symptoms
are present, they most prominently include a significant degree of fatigue out
of proportion to what would be expected for either the severity of the liver
disease or the age of the patient.
Pruritus is seen in approximately 50% of patients at the
time of diagnosis, and it can be debilitating. It might be intermittent and
usually is most bothersome in the evening.
Pruritus that presents prior to the development of
jaundice indicates severe disease and a poor prognosis.
Physical examination can show jaundice and other
complications of chronic liver disease, including hepatomegaly, splenomegaly,
ascites, and edema.
Other features that are unique to PBC include
hyperpigmentation, xanthelasma, and xanthomata, which are related to the
altered cholesterol metabolism seen in this disease.
Hyperpigmentation is evident on the trunk and the arms
and is seen in areas of exfoliation and lichenification associated with
progressive scratching related to the pruritus.
Bone pain resulting from osteopenia or osteoporosis is
occasionally seen at the time of diagnosis.
32. Pathogenesis of basic syndromes
in liver cirrhosis (portal hypertension, ascites, hepatic encephalopathy.

Pathogenesis of basic syndromes in liver
cirrhosis-
-Liver cells regenerate in an abnormal
pattern primarily forming nodules that are surrounded by fibrous tissue.
In these nodules, regenerating hepatocytes
are disorderly disposed. Biliary tract, central vein and the radial pattern of
hepatocytes are absent.
Grossly abnormal liver architecture
eventually ensues that can lead to decreased blood flow to and through the
liver.
Decreased blood flow to the liver and blood
back up in the portal vein and portal circulation leads to some of the serious
complications of cirrhosis.
those are portal hypertension, hepatic
encephalopathy and ascites.
Portal hypertension-
svenous compression and/or obstruction leading
to an increase in vascular resistance.
in cirrhosis, it occurs at the level of the
sinusoid and affects the hepatic microcirculation. In a cirrhotic liver,
on‐going fibrogenesis results in the release of a number of endogenous factors
such as endothelin‐1, α‐adrenergic stimulus and angiotensin II.
These factors cause vasoconstriction and
increased vascular resistance. In addition, production of nitric oxide, a
vasodilator, is reduced.
-Ascites due to portal hypertension. In case of portal hypertension the
abnormality in metaboilc processes occur. This leads to the decreasing of the
albumin level in blood and cause ascite and edema.
Development of ascites in cirrhosis:

-encephalopathy
Cirrhosis, especially in advanced cases, can
cause profound abnormalities in the brain. In cirrhosis, some blood leaving
(eg-ammonia )the gut bypasses the liver as blood flow through the liver is
decreased.
Metabolism of components absorbed in the gut
can also be decreased as liver cell function deteriorates. Both of these
derangements can lead to hepatic encephalopathy as toxic metabolites ammonia ,
normally removed from the blood by the liver, increased ammonia level in blood
and this can reach the brain.In severe cases, hepatic encephalopathy can lead
to stupor, coma, brain swelling and death.
33. The major symptoms (describe
the clinical syndromes) of liver cirrhosis. Instrumental and laboratory methods
of diagnostics.
Clinical picture-
-Cutaneous
manifestations of cirrhosis include: jaundice, spider naevi, petechial skin
rash, palmar erythema, leuconychia (“white nails”) and finger clubbing.
Males may develop gynecomastia and impotence.
Females may have breast atrophy, irregular
menses and amenorrhoea. Loss of axillary and pubic hair is noted in both men
and women.
Patient may have edemas of lower extremities
(soft and warm). Evidence of hepatic encephalopathy becomes increasingly common
with advancing disease.
The person's breath may have a musty sweet
odor (fetor hepaticus).Tongue is bright red color (crimson), with atrophy of
the papillae.
Palpation of the abdomen. Ascites and Caput
Medusa develop in portal hypertension. Hepatomegaly is common in cirrhosis. The
liver is often hard, irregular, and painless. Later liver may decreased in a
size.
Splenomegaly and hypersplenism occurs in more
advanced disease. In patients with
previously stable cirrhosis, decompensation may occur due to various causes,
such as constipation, infection (of any source), increased alcohol intake,
medication, bleeding from esophageal varices or dehydration.
Diagnosis-
Blood test- Blood test may reveal anemia, leukopenia, thrombocytopenia,
markedly increased ESR level.
Biochemical blood test- Aminotransferases - AST and ALT are moderately elevated.
However, normal aminotransferases do not exclude cirrhosis.
Alkaline phosphatase ,usually slightly
elevated. Gamma-glutamyltranspeptidase ,correlates with alkaline phosphatase
levels. It is typically much higher in alcoholic chronic liver disease.
Bilirubin , may elevate as cirrhosis
progresses. Albumin - levels fall as the synthetic function of the liver
declines with worsening cirrhosis since albumin is exclusively synthesized in
the liver.
Globulins ,increased due to shunting of
bacterial antigens away from the liver to lymphoid tissue.
Serum sodium , hyponatremia due to inability
to excrete free water results from high levels of ADH and aldosterone.
Prothrombin time - increases since the liver synthesizes clotting factors.
Blood tests that check for
conditions -that may cause cirrhosis include.
Antinuclear antibodies (ANA) - testing
anti-smooth-muscle antibody (ASMA) testing
(may help detect the presence of autoimmune
chronic hepatitis. )
Anti mitochondrial antibody test (AMA) (which
may help detect primary biliary cirrhosis.)
Ferritin and iron tests(which may help
diagnose iron overload, or hemochromatosis)
Virus antibody testing for hepatitis B, C or
tests for hepatitis virus genetic material (RNA or DNA) (which may help
diagnose infection with certain hepatitis viruses. )
Blood alcohol level testing( These tests may
detect alcohol use, which can cause alcoholic cirrhosis.)
Serum ceruloplasmin testing (which may help
diagnose Wilsosns disease.)
Alpha1-antitrypsin level. (This may diagnose
a condition in which people lack this protein (alpha1-antitrypsin deficiency).
)
Ultrasound -is routinely used in the evaluation of cirrhosis (where it may
show a small and nodular liver in advanced cirrhosis along with increased
echogenicity with irregular appearing areas.)
cirrhosis in imaging are an enlarged caudate
lobe, widening of the liver fissures and enlargement of the spleen.
An enlarged spleen (splenomegaly), which
normally measures less than 11-12Ԝcm in adults.
is very suggestive of cirrhosis with portal
hypertension in the right clinical setting.
Ultrasound may also screen for hepatocellular
carcinoma, portal hypertension.
FibroScan -(transient elastography), uses elastic waves to determine liver
stiffness which theoretically can be converted into a liver score based on the
METAVIR scale.
(The FibroScan produces an ultrasound image
of the liver (from 20–80Ԝmm) along with a pressure reading (in kPa). The test
is much faster than a biopsy (usually last 2.5-5 minutes) and is completely
painless. It shows reasonable correlation with the severity of cirrhosis.)
Gastroscopy -(endoscopic examination of the esophagus, stomach and duodenum)
is performed in patients with established cirrhosis to exclude the possibility
of esophageal varices.
Liver biopsy- in which a sample of liver tissue is removed and analyzed, also
may be done. It is the only test that can
confirm a diagnosis of cirrhosis. It also helps
determine its cause, treatment possibilities, the extent of damage, and the
long-term outlook.
34. Medical management of liver cirrhosis.
cannot be reversed, but treatment could stop
or delay further progression and reduce complications. A healthy diet is
encouraged.
Antibiotics will be prescribed for
infections, and various medications can help with itching. Laxatives, such as
lactulose, decrease risk of constipation; their role in preventing
encephalopathy is limited.
Patients with decompensated cirrhosis
generally require admission to hospital, with close monitoring of the fluid
balance, mental status, and emphasis on adequate nutrition and medical
treatment - often with diuretics, antibiotics, laxatives and/or enemas,
thiamine and occasionally steroids, acetylcysteine and pentoxifylline.
Administration of saline is generally avoided
as it would add to the already high total body sodium content that typically
occurs in cirrhosis. Liver transplantation.
If complications cannot be controlled or when
the liver ceases functioning, liver transplantation is necessary.
35. Treatment of: portal
hypertension, ascites, encephalopathy, hepatorenal syndrome.
Hypertension-
Managed with propanolol to prevent bleeding.
Vitamin k- because of elevated prothrombin
time
.
Ascites-
Edema and fluid overload in 3rd
space such as ascites are managed with diuretics.
Mostly spironolactone ,which results high
aldesterone state.
Encephalopathy -
Is managed with neomycin or lactulose a nonabsorbed
disaccharide that bacteria metabolize in the colon.making it more acidic it
convert to NH3(not absorbed very well)----> NH4 ion. This is excretion of NH3 from the body.
Hepatorenal syndrome- treatment of hepatorenal syndrome is connected with underlying
liver disease.
Midodrine and alpha-agonist
And octreotide may be beneficial in
hepatorenal syndrome.
Best treatment is liver transplantation.
36. Describe the complications of
liver cirrhosis.
Check Number 32
37. Liver failure (hepatic
insufficiency). Classification. Therapy.
liver failure
is characterised by the sudden loss of hepatic function due to
hepatocyte necrosis resulting in hepatic encephalopathy (HE), jaundice and
coagulopathy.
LF is rare, with an incidence of 1–6
cases/million/year in the Western world, and is associated with a high
mortality.
In the
developing world, viral causes are more common than in the Western world where
drug‐induced liver injury predominates.
Causes-
drugs
Paracetamol (acetominophen) is commonly
implicated in overdoses along with non‐ steroidal anti‐inflammatory drugs
(NSAIDs).
Poisons
Amanita species
Metabolic
Acute willson’s
Viral
Hepatitis A,B and E
Adeno virus, HSV/EBV
Others
Pregnancy associated liver disease
Infiltrative eg: lymphoma
classification-
Hyper acute
|
Acute
|
Subacute
|
|
Time from jaundice to HE
|
0-7 days
|
1-4 weeks
|
4-12 weeks
|
Typical cause
|
Paracetamol
|
Hepatitis A,B and E
|
Drugs(non paracetamol)
|
Jaundice
|
mild
|
moderate
|
severe
|
Coagulopathy
|
Severe
|
moderate
|
mild
|
Therapy-
Few specific therapies are available. N‐acetylcysteine
(NAC) is effective for paracetamol‐induced hepatoxicity. For patients with
non‐paracetamol‐induced ALF, NAC is also recommended. In cases of acute HBV or
reactivation or flare of HBV, treatment with antivirals is recommended.
Patients with liver failure should be managed
in a HDU or ITU setting with early discussion with the local transplant centre.
-Cardiovascular: Assessment of volume status.
Intravascular volume depletion is common. LF has similar characteristics to
septic shock (i.e. low BP, peripheral vasodilatation, hyperdynamic circulation;
↑ heart rate, ↓ systemic vascular resistance, ↑ cardiac output). Central venous
and arterial catheters may be required. The use of inotropic agents (e.g.
norepinehrine) is common.
-Respiratory: Is the patient able to protect
their airway? Elective intubation is recommended in those with GCS <8 or
severe agitation/confusion (i.e. grade 3 encepholopathy). Assessment of
arterial blood gases. Respiratory complications include pneumonia, aspiration
and acute lung injury.
-Neurological: What is the GCS? Neurological
dysfunction is common but can be subtle with rapid onset. Once the GCS is <8
the patient will have established brain oedema and will deteriorate rapidly.
Once the patient has become agitated or
develops grade 1 or 2 HE or GCS drops to 12, they should be immediately
transferred to liver transplant centre. They should be intubated pre‐transfer.
Patients with high grade HE (>grade 3), young patients (<35 years) and
those with a high serum ammonia level (>150 μmol/L) are at increased risk of
developing cerebral oedema.
Seizures are a poor prognostic sign.
Intracranial monitoring may be required.
-Renal and electrolytes: Renal dysfunction is
common (>50%). Nephrotoxic drugs should not be given. Adequate volume
resuscitation is required. The use of renal replacement therapy, if required,
should be commenced early and is usually continuous veno‐venous haemodialysis.
Hypoglycaemia is common. ↑ Lactate reflects
tissue hypoxaemia. Metabolic acidosis is common. Tight glucose control (6–10
mmol/L) is recommended. Avoid solutions that contain lactate (e.g. Hartmann’s).
Patients are often intravascular depleted.
-Gastrointestinal: ALF is a hypercatabolic
state and nutritional support is important.
-
Haematology: ↑ INR, ↓ platelets, ↓
fibrinogen. Correction of coagulopathy (fresh frozen plasma, platelets,
cryoprecipitate) required only if there are signs of bleeding.
-Infection: A comprehensive septic screen is
recommended in all ALF patients due to the increased susceptibility to
bacterial and fungal infections. Prophylactic antibiotics and antifungals
are recommended. Universal precautions are integral to avoid nosocomial
infections.
38. Metabolic liver disease:
etiology and pathogenesis of alcohol liver disease. Features of treatment.

Alcohol is metabolised mainly in
the liver through 2 main pathways: alcohol dehydrogenase and cytochrome P-450
2E1. In the liver, alcohol is metabolised to acetaldehyde by alcohol
dehydrogenase, which then is metabolised to acetate by the mitochondrial enzyme
acetaldehyde dehydrogenase. Alcohol dehydrogenase and acetaldehyde
dehydrogenase reduce nicotinamide adenine dinucleotide (NAD) to NADH (reduced
form of NAD). Excessive NADH in relation to NAD inhibits gluconeogenesis and
increases fatty acid oxidation, which in turn promotes fatty infiltration in
the liver.
The cytochrome P-450 2E1 pathway
also metabolises alcohol, and generates free radicals through the oxidation of
NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) to NADP.
Chronic alcohol use upregulates cytochrome P-450 2E1 and produces more free
radicals.
Chronic alcohol exposure also
activates a third site of metabolism: hepatic macrophages, which produce
TNF-alpha and induce the production of reactive oxygen species in the
mitochondria.
Alcoholic people are usually
deficient in antioxidants, such as glutathione and vitamin E. Therefore,
oxidative stress promotes hepatocyte necrosis and apoptosis in these patients.
Free radicals can also induce lipid peroxidation, which can cause inflammation
and fibrosis. The alcohol metabolite acetaldehyde, when bound to cellular
protein, produces antigenic adducts and induces inflammation. Alcohol also
affects the barrier function of intestinal mucosa, producing endotoxaemia,
which leads to hepatic inflammation.

Biomedical and cellular pathogenesis of liver injury secondary to chronic ethanol ingestion.
39. Metabolic liver disease:
classification, etiology and pathogenesis of non-alcoholic fatty liver disease
(hepatosis). Principles of treatment.

Non-alcoholic fatty liver disease (NAFLD) is a very common disorder and refers to a group of conditions where there is accumulation of excess fat in the liver of people who drink little or no alcohol. The most common form of NAFLD is a non serious condition called fatty liver.
etiology and pathogenesis
NAFLD is part of the metabolic syndrome characterized by diabetes, or pre-diabetes (insulin resistance), being overweight or obese, elevated blood lipids such as cholesterol and triglycerides, as well as high blood pressure. Not all patients have all the manifestations of the metabolic syndrome. Less is known about what causes NASH to develop. Researchers are focusing on several factors that may contribute to the development of NASH. These include:
- Oxidative stress (imbalance between pro-oxidant and anti-oxidant chemicals that lead to liver cell damage)
- Production and release of toxic inflammatory proteins (cytokines) by the patient’s own inflammatory cells, liver cells, or fat cells
- Liver cell necrosis or death, called apoptosis
- Adipose tissue (fat tissue) inflammation and infiltration by white blood cells
- Gut microbiota (intestinal bacteria) which may play a role in liver inflammation
treatment.
- Weight reduction (diet + exercise, medications, surgery)
- Lipid lowering medications
- Insulin sensitizers (medications)
- Decrease the amount of liver inflammation by administering anti-oxidant medications, anti-apoptotic medications and anti-cytokine medications
40. Disorders of bilirubin
metabolism leading to hyperbilirubinemia (Gilbert’s, Crigler-Najjar’s, Rotor’s,
Dubin-Johnson’s syndromes).
41. Hemochromatosis. Etiology.
Pathogenesis. Symptoms and methods of diagnostics. Principles of treatment.
MRI can eliminate the need for biopsy if both are present.
Etiology-
HH is genetic disorder of unregulated iron
absorption, leading to a state of iron overload.
Excess iron is deposited in organs, such as
the liver, pancreas, pituitary, skin and joints, which over many years leads to
organ damage and dysfunction.
The highest incidence of HH worldwide is seen
in Ireland. It is infrequently encountered in southern Europe, Africa and Asia.
Pathogenesis-
In this disease there is an overabsorption of
iron in the duodenum, leading to iron buildup in a number of tissues throughout
the body.
This leads to chronic hepatic inflammation
and fibrosis.
Clinical presentation-
Cardiomyopathy develops in 15% of the
patient.
Arthralgias ,skin hyperpigmentation,
diabetes,hypogonadism are also common.
Vibrio vulnificus and yersinia infections
occur with increased frequency because of their avidity for iron.
Diagnosis-
In serum test-
Elevated iron level and diminished iron
binding capacity (>300 micro gram /l in men and after menopausal women , in
young women >200 micro gram /l)
Ferritin also elevated.
Liver biopsy is the most accurate test and
abnormal C282Y gene.
MRI can eliminate the need for biopsy if both are present.
Treatment-
Phlebotomy is used to remove large ammount of
iron from body.
Deferoxamine and deferasirox are used only in
those who cannot undergo phlebotomy.
42. Wilson's disease. Etiology.
Pathogenesis. Symptoms and methods of diagnostics. Principles of treatment.
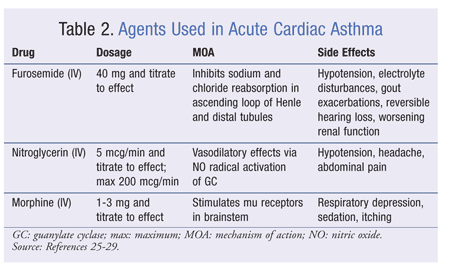



Etiology-
An autosomal recessive disorder that leads to
impaired cellular transport of copper and accumulation in the liver, brain and
cornea. Affects 1 in 30 000 live births. Occurs due to mutations of the ATP7B
gene on chromosome 13. Variable clinical penetrance with presentation in
childhood or adulthood. Majority of patients present between the ages of 5 and
35 years.
Pathogenesis-
This is an autosomal recessive
disorder,leading to the diminished ability to excrete copper from the body and
also increased absorption of the copper from the small intestine.
Clinical picture-
Copper build up in brain,liver and cornea.
Basal ganglia dysfunction contribute to the
movement disorder that develops.
10% of the patient have the psychiatric
disturbance.
Kayser-fleischer rings (blue -grey color
lines) are found in the eye on slit lamp.
Tremor and parkinson’s occur in 1/3 of
patients.
Fanconi syndrome (glucosuria,amino aciduria
,hypouricaemia and proximal renal tubular acidosis) and type 2 proximal renal
tubular acidosis develop in the kidney.
Jaundice , steatosis ,acute liver failure.
Diagnosis-
Wilson’s disease should be considered in any
patient with unexplained liver, neurologic or psychiatric symptoms.
• Slit‐lamp examination: examine for KF
rings.
• Serum ceruloplasmin: decreased by 50% but
can be normal.
• Serum copper: low. Can be high in patients
with cholestasis.
• 24‐hour urinary copper: >1.6 μmol/24
hours. Can be high in patients with an acute hepatitis or with cholestasis.
Raised by penicillamine in affected patients more than carriers.
• Liver biopsy: parenchymal copper >4
μmol/g dry weight. Changes include steatosis to macronodular cirrhosis.
• Mutation analysis: whole gene sequencing is
possible. Recommended for screening first‐degree relatives of patients with
Wilson’s disease.
Treatment-
1)D‐penicillamine: Promotes urinary excretion
of copper and induces metallothionein (endogenous chelator of metals). Usually
taken 1–2 hours before meals to inhibit dietary absorption of copper.
Adequacy of treatment can be monitored by
performing 24‐hour urinary copper excretion.
Goal of therapy is to increase urinary copper
excretion from baseline levels and a normal serum copper.
2)Trientine: Chelates copper and then promotes urinary copper
excretion. Associated with iron deficiency. Fewer side effects than
D‐penicillamine.
3)Zinc: Reduces uptake of copper from the gastrointestinal tract and
induces metallothionein in enterocytes. Bound copper is then excreted faecally.
4)Liver transplantation: Medical therapy is
rarely effective in patients with acute liver failure.
43. Functional biliary disorders
(gallbladder and Oddi's sphincter dysfunction). Diagnostic criteria. Therapy
depending on a form of the dysfunction.
Functional gallbladder disorder
is defined as biliary pain resulting from a primary gallbladder motility
disturbance in the absence of gallstones, sludge, microlithiasis, or
microcrystal disease. The diagnosis is considered in patients with typical
biliary-type pain who have had other causes for the pain excluded.
Following removal of
the gallbladder, biliary pain has been attributed to sphincter of Oddi
dysfunction (SOD).
SOD represents intermittent obstruction to the flow of biliopancreat‐
ic secretions
through the sphincter of Oddi in the absence of biliary stones or a ductal
stricture
Clinical criteria —
Consensus guidelines (the Rome IV criteria) have been developed to help diagnose
functional gallbladder disorder. Patients who fulfill these criteria should
undergo an evaluation for functional gallbladder disorder, whereas patients who
do not fulfill all of the criteria should be evaluated for alternative causes
of their abdominal pain.
Rome IV criteria for
functional gallbladder disorder require:
●Biliary pain
●Absence of
gallstones or other structural pathology
In addition, the
criteria that are supportive of functional gallbladder disorder, but are not
required, include:
●Low ejection
fraction on scintigraphy
●Normal liver
enzymes, conjugated bilirubin, and amylase/lipase
To fulfill the Rome
IV criteria for biliary-type pain, patients need to have pain that:
●Is located in the
epigastrium and/or right upper quadrant
●Occurs at variable
intervals (not daily)
●Lasts at least 30
minutes
●Builds up to a
steady level
●Is severe enough to
interrupt daily activities or lead to an emergency department visit
●Is not significantly
(<20 percent) relieved by bowel movements, postural changes, or acid
suppression
Criteria that are
supportive of biliary pain, but are not required, include: (a) pain that is
associated with nausea and vomiting, (b) pain that radiates to the back and/or
right subscapular region, and (c) pain that awakens the patient from sleep in
the middle of the night.
MANAGEMENT — Cholecystectomy is
the treatment for functional gallbladder disorder. Patients are candidates for
cholecystectomy if they fulfill the clinical criteria for functional
gallbladder disorder, if alternative explanations for their symptoms have been
excluded, and if their gallbladder ejection fraction (GBEF) is reduced (<40
percent).
Alternative
therapies, such as bile acid composition modifiers, promotility agents, and
anti-inflammatory drugs, have not been adequately studied
1. Emergency care in pulmonary hemorrhage.
Immediate treatment
of P-Hem should include tracheal
suction, oxygen and
positive pressure ventilation. To assist in decreasing P-Hem, mean
airway pressure
should be increased, either by a relatively high PEEP Positive End
Expiratory Pressure cmH2O) or by high
frequency ventilation. Correct underlying abnormalities, especially
disorders of
coagulation. When blood loss is large, prompt blood transfusion may be
needed to maintain
an adequate circulating blood volume. The outcome is dependent on
the cause of P-Hem.
Mortality is 30 to 40%.
2. Emergency treatment in patients with attack of bronchial asthma.
Oxygen: Administration of
oxygen through nasal cannulae or a mask is recommended to maintain SaO2 at
greater than 90% (> 95% in pregnant women and patients with concomitant
heart disease).
Inhaled
short-acting β2-agonists: 10 mg of salbutamol
Corticosteroids: Continue
prednisolone 40-50 mg daily for at least five days or until recovery
Ipratropium bromide: Add nebulised ipratropium bromide (0.5 mg 4- to 6-hourly)
- Consider giving an infusion of IV magnesium sulfate (only after consultation with senior medical staff) for patients with life-threatening or near-fatal asthma, or with acute severe asthma who have not had a good initial response to inhaled bronchodilator therapy.
Antibiotics are not routinely indicated.
3. Emergency treatment in uncomplicated hypertensive crisis.
Captopril (sublingual)
Nifedipine
Nitroglycerin
Monoxidine
Beta blockers
4. Emergency treatment in complicated hypertensive crisis.
IV infusions only
Nitroglycerin
labetalol
Enalaprilat
Clonidine
Hydralazine
Esmolol
5. Emergency care of patients with unstable angina.
Aspirin 525mg chewing
clopidlogel 300 - 600 mg chewing
beta blockes
Morphine IV start from 1mg and increase. nitric oxide can be used in place of morphine
6. Analgesia in myocardial infarction patients.
Morphine IV start from 1mg and increase
7. Emergency therapy of cardiogenic shock.
- Initial stabilization. If there is a high flow of O2, endotracheal intubation, intravenous access, cardiac rhythm, and pulse oximetry monitoring should be considered.
- Rhythm disturbances, hypoxemia, hypovolemia, and electrolyte abnormalities should be identified and treated.
- Aspirin 160 to 325 mg. The patient should chew and swallow unless there is an allergy or contraindication.
- Intravenous nitroglycerin or morphine sulfate, titrated to response, should be administered as needed for chest pain, as well as hemodynamic parameters monitored.
- For mild to moderate hypotension, in absence of hypovolemia, dobutamine 2.5 to 20.0 µg/kg per min should be administered. For severe hypotension dopamine 2.5 to 20.0 µg/kg per min should be used, titrated to desired effect and utilized at the lowest dose possible.
- Intravenous nitroglycerin 5 to 100 µg/min and sodium nitroprusside 0.5 to 10.0 µg/kg per min should be used to improve cardiac output through reduction of preload and afterload.
- Intra-aortic balloon pump. To decrease afterload the measure should be temporized and diastolic pressure and coronary perfusion augmented.
- Thrombolytic therapy, percutaneous transluminal angioplasty (PCTA), emergent CABG should be used as indicated or available.
- Cardiology and cardiac surgery should be consulted early. Transfer should be arranged if indicated.
8. Emergency care for patients with attack of heart asthma.
9. Emergency care in pulmonary edema.
10. Emergency care in the paroxysm of atrial fibrillation.
- The management of AF involves control of the arrhythmia (by rhythm or rate control) and thromboprophylaxis to prevent strokes.
- Treat any underlying cause - eg, acute infection, hyperthyroidism. AF may revert on treatment or resolution of an associated problem - eg, acute infection or alcohol intoxication.
- Treat associated heart failure.
11. The urgent aid in patients with a paroxysm of supraventricular tachycardia.

12. Emergency therapy in patients with a paroxysm of ventricular tachycardia.
Direct current cardioversion is recommended for patients presenting with sustained VT and haemodynamic instability.
In patients presenting with sustained haemodynamically tolerated VT in the absence of structural heart disease (e.g. idiopathic RVOT), i.v. flecainide or a conventional beta-blocker, verapamil or amiodarone may be considered.
Direct current cardioversion is recommended for patients presenting with sustained VT and haemodynamic instability.
In patients presenting with sustained haemodynamically tolerated VT in the absence of structural heart disease (e.g. idiopathic RVOT), i.v. flecainide or a conventional beta-blocker, verapamil or amiodarone may be considered.
13. The urgent aid in patients with ventricular asystole.

14. The urgent aid in patients with ventricular fibrillation and flutter.
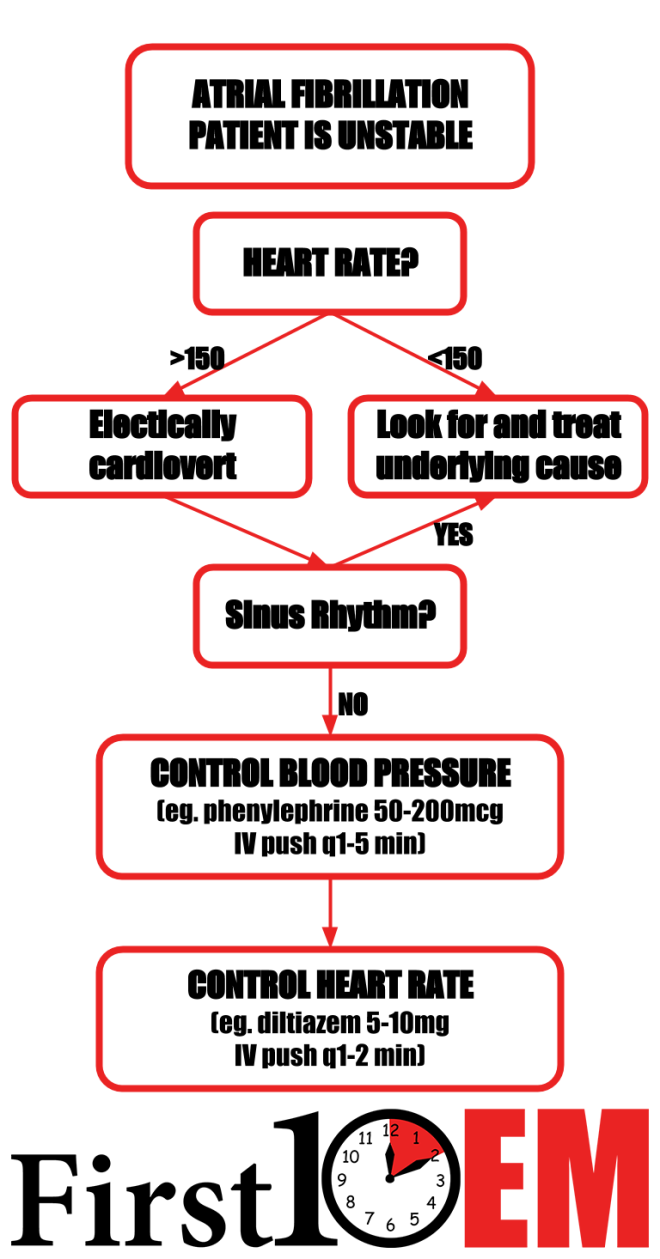
15. Emergency aid for patients with onset of Morgagni-Adams-Stokes.
Stokes-Adams syndrome was the chief impetus for development of a device that monitors and regulates the heartbeat (artificial pacemaker) and these have been in use for over 30 years. Surgical implantation of a pacemaker into the heart is still the treatment of choice. In emergency situations, anticholinergics or sympathomimetics may be prescribed temporarily while a temporary pacemaker is inserted until a permanent pacemaker can be implanted.
Stokes-Adams syndrome was the chief impetus for development of a device that monitors and regulates the heartbeat (artificial pacemaker) and these have been in use for over 30 years. Surgical implantation of a pacemaker into the heart is still the treatment of choice. In emergency situations, anticholinergics or sympathomimetics may be prescribed temporarily while a temporary pacemaker is inserted until a permanent pacemaker can be implanted.
16. Emergency aid for patients with gastrointestinal bleeding.
- Correct fluid losses (place two wide-bore cannulae and also send bloods at the same time). Either colloid or crystalloid solutions may be used to achieve volume restoration prior to administering blood products; red cell transfusion should be considered after loss of 30% of the circulating volume.[3]
- Transfuse patients with massive bleeding with blood, platelets and clotting factors in line with local protocols for managing massive bleeding. Major haemorrhage protocols should be in place.[3]
- Decisions on blood transfusion should be based on the full clinical picture; over-transfusion may be as damaging as under-transfusion.[6][7]
- Platelet transfusions should not be offered to patients who are not actively bleeding and are haemodynamically stable.
- Platelet transfusions should be offered to patients who are actively bleeding and have a platelet count of less than 50 x 109/L.
- Fresh frozen plasma should be used for patients who have either a fibrinogen level of less than 1 g/L, or a prothrombin time (INR) or activated partial thromboplastin time greater than 1.5 times normal.
- Prothrombin complex concentrate should be used for patients who are taking warfarin and actively bleeding.
- Recombinant factor Vlla should not be used except when all other methods have failed.
Recommendations emphasise early risk stratification, using validated prognostic scales, and early endoscopy (within 24 hours). The following formal risk assessment scores are recommended by the National Institute for Health and Care Excellence (NICE) for all patients with acute UGIB:
17. Emergency aid for patients with gastrointestinal bleeding from varices of esophagus.
18. Emergency care in patients with anaphylactic shock.
Awesome
ReplyDeleteHere is a great herbal doctor who cured me of Hepatitis B. his name is Dr. Imoloa. I suffered Hepatitis B for 11 years, I was very weak with pains all over my body my stomach was swollen and I could hardly eat. And one day my brother came with a herbal medicine from doctor Imoloa and asked me to drink and I drank hence there was no hope, and behold after 2 week of taking the medicine, I started feeling relief, my swollen stomach started shrinking down and the pains was gone. I became normal after the completion of the medication, I went to the hospital and I was tested negative which means I’m cured. He can also cure the following diseases with his herbal medicine...lupus, hay fever, measles, dry cough, diabetics hepatitis A.B.C, mouth ulcer, mouth cancer, bile salt disease, fol ate deficinecy, diarrhoea, liver/kidney inflammatory, eye cancer, skin cancer disease, malaria, chronic kidney disease, food poisoning, parkinson disease, bowel cancer, bone cancer, brain tumours, asthma, arthritis, epilepsy, cystic fibrosis, lyme disease, muscle aches, fatigue, alzhemer's disease, acute myeloid leukaemia, acute pancreatitis, chronic inflammatory joint disease, Addison's disease back acne, breast cancer, allergic bronchitis, Celia disease, bulimia, congenital heart disease, cirrhosis, constipation, fungal nail infection, fabromyalgia, (love spell) and many more. he is a great herbalist man. Contact him on email; drimolaherbalmademedicine@gmail.com. You can also reach him on whatssap- +2347081986098.
ReplyDeleteVery impressive article! The blog is highly informative and has answered all my questions. To introduce about our company and the activities, E-Healthcare Lists
ReplyDeleteis one of the global suppliers of healthcare mailing list & email list. Want to begin your business with the Gastroenterologists? To sign the vital deals, you need an authentic
Gastroenterologists mailing list.